Original Breve
Metabolismo mineral óseo en pacientes con enfermedad renal crónica: Revisión sobre su fisiopatología y morbimortalidad
Bone mineral metabolism in patients with chronic kidney disease: Review of its pathophysiology and morbimortality
Jeanette Bernuy1,a, Gustavo F. Gonzales2,b
1 Hospital Nacional Cayetano Heredia y Facultad de Medicina Universidad Peruana Cayetano Heredia. Lima, Perú.
2 Laboratorio de Endocrinología y Reproducción Facultad de Ciencias y Filosofía, Universidad Peruana Cayetano Heredia. Lima, Perú.
a Médico nefróloga; b biólogo y médico endocrinólogo; doctor en Medicina y doctor en Ciencias.
RESUMEN
La enfermedad mineral ósea (EMO) es un término amplio que incluye a las alteraciones séricas del calcio, fósforo, vitamina D, paratohormona, anormalidades en el crecimiento, mineralización ósea y/o a las calcificaciones extraesqueléticas que acompañan al paciente con enfermedad renal crónica (ERC). Está presente en casi la totalidad de pacientes en diálisis y con el trasplante renal puede no siempre mejorar. Se han identificado nuevos factores y hormonas; como klotho y factor de crecimiento de fibroblastos-23 (FGF-23) que interactúan con la vitamina D y con la paratohormona en el manejo renal del calcio y fósforo. Ciertos reportes indican que son marcadores precoces del desarrollo de EMO, incluso cuando la función renal está levemente disminuida y los niveles de paratohormona son normales. La EMO ha sido asociada con mayor mortalidad, principalmente por su vinculación con la calcificación vascular. Este proceso conlleva a un incremento de eventos cardiovasculares que constituyen la principal causa de morbimortalidad en pacientes con ERC, sobre todo aquellos que se encuentran en diálisis, independientemente de la modalidad que los pacientes sigan. La forma de presentación de la EMO puede ser de alto o bajo recambio. Aunque no está completamente definido qué es lo que determina que se exprese una en particular, se ha encontrado que la enfermedad de bajo recambio se relaciona con malnutrición, uso inadecuado de calcitriol y diálisis ineficiente. El conocimiento de la EMO es relevante por su asociación con las complicaciones mencionadas y porque constituye un parámetro para evaluar la terapia instalada.
Palabras clave: Hormona paratiroidea; Deficiencia de vitamina B; Enfermedad renal crónica; Trastornos del metabolismo del calcio (fuente: DeCS BIREME).
ABSTRACT
Mineral Bone Disorder (MBD) is a broad term that includes abnormal serum calcium, phosphorus, vitamin D, parathyroid hormone, growth abnormalities, bone mineralization and/or extraskeletal calcifications in patients with chronic kidney disease (CKD ). It is present in almost all patients on dialysis and may not always improve with a kidney transplant. New factors and hormones have been identified, such as Klotho and fibroblast growth factor-23 (FGF-23) that interact with vitamin D and the parathyroid hormone in the renal management of calcium and phosphorus. Some reports indicate that they are early markers of the development of MBD, even when kidney function is slightly decreased and parathyroid hormone levels are normal. MBD has been associated with higher mortality, mainly because of its link with vascular calcification. This process leads to an increase in cardiovascular events which are the leading cause of morbidity and mortality in CKD patients, especially those who are on dialysis, regardless of the modality that the patients follow. The presentation of the BMD can be of high or low turnover. Although it is not completely defined what determines that a particular form of presentation is expressed, it has been found that the low turnover disease is related to malnutrition, inappropriate use of calcitriol and inefficient dialysis. Knowledge of BMD is relevant for its association with the complications mentioned above and because it constitutes a parameter for assessing the instituted therapy.
Key words: Parathyroid hormone; Vitamin B deficiency; Chronic renal disease; Calcium metabolism disorders (source: MeSH NLM).
INTRODUCCIÓN
La enfermedad mineral ósea (EMO) se observa en pacientes con enfermedad renal crónica (ERC) y se define como cualquier anormalidad en el metabolismo del calcio, fósforo, paratohormona o vitamina D, anormalidad en el recambio óseo, mineralización, fuerza, crecimiento lineal y/o presencia de calcificaciones extraesqueléticas asociada a la enfermedad renal. El término osteodistrofia que antiguamente se empleaba para expresar el efecto de la ERC a nivel óseo está limitado a las alteraciones histomorfométricas en pacientes con enfermedad renal crónica (1).
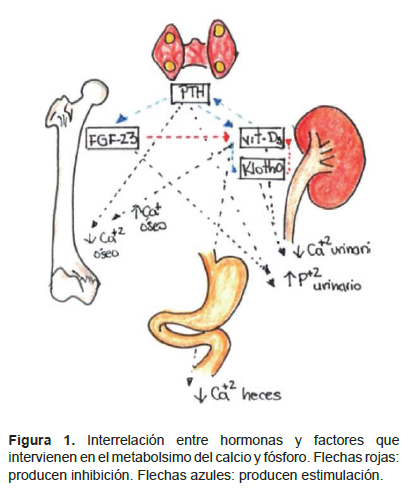
Esta condición se describió desde los años 50 en aquellos pacientes cuya tasa de filtración glomerular disminuía por debajo de los 60 mL/min. Casi la totalidad de pacientes que se encuentren en el estadio 5 de enfermedad renal crónica (depuración menor de 15 mL/min) cursarán con algún grado de EMO. Con la pérdida de la masa renal, la síntesis de calcitriol disminuye y, por tanto, la capacidad renal de promover la reabsorción de calcio se va perdiendo. Adicionalmente, la función paratiroidea se incrementa en el afán de normalizar los niveles séricos de calcio y fósforo. Otros factores, proteínas y hormonas han sido descritos como el FGF-23, klotho, entre otros que interactúan favoreciendo la excreción de fósforo y reabsorción de calcio (2-4).
La ERC es una enfermedad emergente, considerando el hecho de que las dos principales etiologías son: diabetes mellitus e hipertensión arterial, enfermedades que van en aumento, así como ocurre con el envejecimiento poblacional. Dado que una de las complicaciones de la ERC es la enfermedad mineral ósea y que esta se relaciona con los eventos cardiovasculares, que es causa de muerte en los pacientes con ERC, se hace necesario conocer su fisiopatología para poder brindar una orientación a los pacientes que están en riesgo, a los que la padecen y saber referirlos oportunamente al médico especialista.
El objetivo del presente artículo es revisar factores recientemente asociados al metabolismo del calcio y fósforo, haciendo relevancia en los distintos cambios que ocurren con la pérdida de la función renal. Para ello se ha realizado una revisión narrativa o no sistemática de la literatura. Asimismo, se destaca el efecto de dichas alteraciones en la calcificación vascular. Finalmente, se comenta las variaciones metabólicas que ocurren luego del trasplante renal.
FACTORES RELACIONADOS AL DESARROLLO DE ENFERMEDAD MINERAL ÓSEA
Se dice que hay diferencias raciales en las manifestaciones de la EMO. En individuos afroamericanos con ERC se ha encontrado mayor densidad mineral, mayor tasa de formación ósea, mayor volumen óseo, niveles más elevados de PTH y menores niveles de fósforo, calcio y FGF-23 que la población caucásica americana con ERC (5,6).
Se ha descrito diferencias en los niveles de fósforo y FGF-23 según el sexo, donde la población femenina es la que cursa con mayor frecuencia la osteopenia y la osteoporosis (7-9). El grado de severidad de la EMO depende también del grupo etario al cual pertenece el paciente, por ejemplo, la población pediátrica cursa con retardo en el crecimiento, menor estatura y osteodistrofia renal, producto de una combinación de anormalidades en la hormona del crecimiento, deficiencia de vitamina D, hiperparatiroidismo, hipogonadismo y nutrición inadecuada, siendo el impacto de la enfermedad mineral ósea mediato y a largo plazo (10). Niños más pequeños con ERC cursan con reducciones significativas de aéreas corticales y trabeculares óseas (11).
Se revisan los factores adicionales a vitamina D y paratohormona que cumplen una función importante en el manejo renal del calcio y fósforo.
FACTOR DE CRECIMIENTO DE FIBROBLASTOS 23 (FGF-23)
El FGF-23 es producido principalmente por los osteocitos y osteoblastos, y se considera un inhibidor de la mineralización. Fue identificado por Yamashita en el año 2000, detectado en el núcleo talámico ventrolateral de embriones de ratones (12).
En el año 2004 se encontró que en pacientes con raquitismo hipofosfatémico autosómico dominante (ADHR) existía una mutación sin sentido en el gen del FGF-23, lo cual otorgaba resistencia para su proteólisis y, por tanto, sus funciones permanecían activas y llevaban al desarrollo de dicha enfermedad (13). Adicionalmente, se aisló FGF-23 de tumores que ocasionaban hipofosfatemia y, consecuentemente, osteomalacia debido a pérdida renal de fosfato (osteomalacia inducida por tumores: TIO) (14). El FGF-23 se identificó inicialmente en osteoblastos y osteocitos, pero también ha sido descrito en glándulas salivales, estómago, músculo esquelético, cerebro, hígado, corazón y glándula mamaria (15).
El FGF-23 pertenece a una gran familia cuyo ancestro es el FGF-13. Los FGF se dividen en siete subfamilias filogenéticas, cada una de ellas compuesta por tres grupos. En general, la mayoría de los FGF con función paracrina requieren de la presencia de heparin sulfato y glucosaminoglicanos para permitir una señal de transducción y ejercer roles en: crecimiento tumoral, desarrollo embrionario, angiogénesis y cicatrización de heridas. Sin embargo, la familia FGF-19 que incluye al FGF-19, FGF-21 y FGF-23, tiene una afinidad reducida por heparin sulfato, esto limita su permanencia en la matriz extracelular y que tengan por tanto una función endocrina. La afinidad reducida al heparin sulfato reduce la capacidad de interacción entre los FGF y sus receptores (FGFR). Es así que en la subfamilia FGF-19 la activación de los FGFR requiere de un cofactor (16,17).
El efecto principal FGF23 es actuar como una hormona contrarreguladora de la vitamina D, reduciendo los niveles de 1,25(OH)2 D al actuar sobre las enzimas Cyp27b1 y Cyp24a1 de tal manera que disminuye su producción y se incrementa su catabolismo (18). El FGF suprime la expresión del cotransportador de sodio y fosfato NPT2a y NPT2c incrementando así la excreción urinaria de fosfato a nivel de túbulo contorneado proximal (18,19).
KLOTHO
Fue identificado en 1997 mientras se estudiaba el fenotipo de ratones que sobreexpresaban el intercambiador Na+/H+ tipo I. Una mutación en una región del promotor, que sería luego identificada como Klotho, ocasionaba un proceso de envejecimiento acelerado. Estos ratones desarrollaron osteoporosis, arterioesclerosis, hipogonadismo hipogonadotrófico, atrofia cutánea, entre otros (20).
El gen Klotho codifica para una proteína transmembrana (αklotho) que se expresa en múltiples tejidos: plexo coroideo, hígado, tejido adiposo, páncreas, gónadas, placenta, colon, vejiga entre otros, pero es particularmente expresado en el riñón. Klotho se expresa de manera importante en túbulos contorneados distales de ratones, ratas y también en el ser humano, ha sido identificado también en el túbulo contorneado proximal y el túbulo colector medular. Pertenece a la familia de las glucosidasas unidas a membrana. Está compuesta por 1014 aminoácidos y su peso molecular es de 130 KDa, el gen se ubica en la porción q12 del cromosoma 13 (21).
También existe una forma soluble que ha sido identificada en varios fluidos corporales como el líquido cefalorraquídeo, la sangre y la orina. La proteína transmembrana y la soluble tienen funciones diferentes.
La fracción de klotho, que es soluble, actuaría como enzima regulando algunas glicoproteínas de superficie. Se ha descrito que puede inhibir a la insulina y al IGF- 1 (la inhibición de éste último ha sido relacionada con mayor sobrevida) (22).
La proteína transmembrana forma un complejo tetramérico con los receptores de FGF (FGFRs) y funciona como cofactor del FGF-23. La acción de klotho sobre la reabsorción de fósforo puede realizar asociado o no a la acción de FGF-23. La porción transmembrana se une al receptor de FGF-23 y así permite la acción fosfatúrica a nivel de los transportadores NaPi2a y NaPi2c. La porción soluble hace que el transportador NaPi2a se vuelva más susceptible a las proteasas y, adicionalmente, inhibe su actividad induciendo la deglicosilación del transportador (23, 24). Además, klotho interviene en la homeostasis del calcio. Imura mostró que klotho estimula la acción del canal de calcio TRPV5 ubicado en el túbulo contorneado distal, estabilizándolo en la superficie celular (25). Chang resalta la función de b glucoronidasa de klotho, evitando la internalización del canal y previniendo así la pérdida renal de calcio (26).
Los canales ROMK1, que permiten la secreción de potasio a nivel renal, también son regulados por klotho pues se ha descrito un efecto similar al que produce con los canales TRPV5. Klotho remueve los ácidos siálicos terminales del canal ROMK1 y de TRPV5 evitando así la endocitosis de los canales transportadores mediados por clatrina (27,28).
El FGF23 reduce los niveles de 1,25(OH)2D al actuar sobre las enzimas Cyp27b1 y Cyp24a1 de tal manera que disminuye su producción y se incrementa su catabolismo. Klotho y vitamina D se regulan recíprocamente. En modelos knock-out para Klotho, se ha observado niveles elevados de vitamina D, con presencia de niveles elevados de 1αhidroxilasa y disminuidos de 24-hidroxilasa. Adicionalmente, se encontró que cuando se alteran los niveles de vitamina D, esto se relaciona con mayor mortalidad en ratones con deficiencia de Klotho y FGF-23. Un incremento en 1,25 Vitamina D3 estimula la expresión de Klotho que suprime, a su vez, la producción de dicha vitamina. klotho regula indirectamente la producción de PTH modulando los niveles de vitamina D activa, fósforo y FGF23. Adicionalmente, existe una acción directa sobre la producción y liberación de PTH pues Klotho se expresa en la membrana de la paratiroides en compañía de FGFR1 y FGFR3 (29).
Se ha encontrado que los niveles de FGF-23 se encuentran elevados en estadios 2 y 3 de ERC. Esta elevación se debería más a un incremento en su producción por los osteocitos que a una acumulación por disminución de la función renal. Esta elevación precede a cualquier incremento detectable en los niveles de fósforo o disminución de los niveles de vitamina D (30,31).
Si bien se han descrito genes cuyas mutaciones se asocian a incremento en la acción de FGF-23, ocasionando enfermedades hereditarias como la hipofosfatemia ligada a X (ocasionada por mutaciones en el gen PHEX), o la hipofostemia recesiva autosómica causada por mutaciones en DMP1 o en ENPP1, en la ERC no se conoce con exactitud cómo los osteocitos censan anormalidades sutiles en el manejo tubular renal del fosfato y si existe o no un gatillo que estimula la liberación de FGF-23 del hueso en respuesta a elevaciones sutiles de fósforo en pacientes con ERC, pues en estudios en voluntarios sanos no hubo un efecto de aumento de los niveles de FGF-23 cuando se incrementó el aporte de fósforo en la dieta (31). Probablemente la elevación precoz de FGF-23 en la ERC y su efecto de reducir la producción renal de vitamina D activa sea un estímulo para el incremento posterior de paratohormona (32).
El FGF-23 ha mostrado reducir directamente la secreción de PTH in vivo e in vitro, esto sugiere que FGF tiene un doble efecto sobre PTH: un efecto indirecto a través de la disminución de la síntesis de vitamina D y un efecto directo sobre las células paratiroideas que expresan diferentes receptores para FGF-23 y para klotho logrando así unirse a FGF23 con gran afinidad. La expresión de estos receptores para FGF-23 y klotho están disminuidos en pacientes con hiperparatiroidismo secundario, lo cual puede explicar porqué FGF-23, incluso en niveles elevados, no puede evitar la elevación de los niveles de PTH (33,34) (Figura 2).
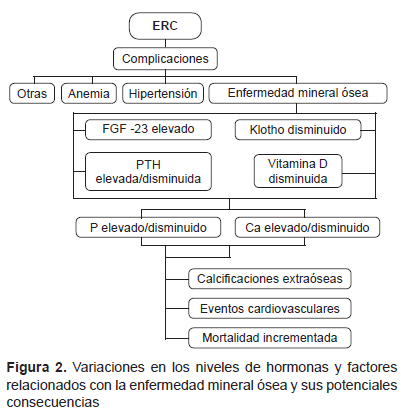
ENFERMEDAD MINERAL ÓSEA Y CALCIFICACIÓN VASCULAR
El FGF-23 ha sido relacionado con complicaciones en diálisis como hipertrofia ventricular izquierda, calcificaciones vasculares y, por tanto, mayor morbimortalidad (35-38). Klotho y FGF-23 tienen efectos en las paredes de los vasos sanguíneos, y disminuye el efecto vasoconstrictor del fosfato y FGF-23 al incrementar la producción de óxido nítrico (39). Kitagawa et al. encuentran que la disminución de la fracción soluble de Klotho está independientemente relacionada con signos de disfunción vascular y rigidez arterial en pacientes con ERC (40).
El proceso de calcificación vascular es complejo, siendo uno de los primeros pasos la diferenciación o transformación de las células del músculo liso de los vasos al genotipo de osteocito o condrocito. Estaría produciéndose una sobrerregulación del factor que se une al core del factor de transcripción alfa-1 (Cbfa-1) más conocido como factor de transcripción-2 relacionado a Runt (RUNX2) (41). Posteriormente, estas células secretan proteínas de matriz. Se ha observado incremento en la expresión de RUNX2 por técnicas de híbridización in situ en áreas adyacentes de calcificación de la íntima y capa media de arteria epigástrica inferior de pacientes en hemodiálisis. Una vez que la matriz se deposita, las células mineralizan la matriz a través de la secreción de vesículas de matriz o por apoptosis. La presencia de calcio y fósforo incrementa el potencial mineralizante de las vesículas de matriz. Esto explicaría por qué en la ERC se observa disturbios del metabolismo mineral y óseo y, concomitantemente, calcificación arterial (42). Tanto en el hueso como en las arterias existen inhibidores de la calcificación que incluyen los pirofosfatos, osteopontinas, así como inhibidores circulantes de la calcificación como fetuina-A (43).
La Fetuin-A es una glicoproteína de 62 kDa sintetizada a nivel hepático. Es miembro de la superfamilia de inhibidores de la cistein proteasa. Es un transportador de factores de crecimiento, que se une al factor de crecimiento transformante-(TGF-b) y a la proteína morfogenética (44,45). Disminuye la activación de macrófagos y la liberación de citokinas proinflamatorias. Interactúa con núcleos minerales para formar partículas coloidales previniendo la precipitación de fosfato de calcio. No afecta la mineralización ósea normal. Adicionalmente, inhibe la apoptosis de las células musculares lisas de los vasos sanguíneos (45).
En modelos animales de ERC los ratones CKD-2 mostraron incremento en la producción de inhibidores de Wnt (wingless/integration) como Dickkopf-1 (Dkk1) y esclerostina y disminución de Klotho soluble(46). La vía Wnt canónica es una vía integrativa que tiene un rol importante en la formación ósea promoviendo la proliferación osteoblástica, la inducción de la osteoblastogénesis, la inhibición de la apoptosis osteocítica y osteoblástica y la atenuación de la osteoblastogénesis (46).
Al neutralizar Dkk1 en el modelo de CKD-2 en ratones, a través de la administración de anticuerpos monoclonales, luego de daño renal, se estimulaba la tasa de formación ósea, se corregía la osteodistrofia y se prevenía la calcificación vascular. Adicionalmente, se suprimía la expresión aórtica del factor de transcripción osteoblástica Runx2, se incrementaba la expresión de la proteína 22a del músculo liso vascular y se restauraba la expresión de klotho en aorta, disminuyendo, a su vez, los niveles de esclerostina (47). Esta glicoproteína ha sido encontrada en la aorta calcificada de humanos y cumpliría una labor de retroalimentación negativa sobre la función osteoblástica, encontrándose una relación con mayor mortalidad en pacientes en diálisis (48,49).
DIÁLISIS Y ENFERMEDAD MINERAL ÓSEA
Aunque algunos estudios no encuentran relevancia clínica entre los valores de FGF y morbimortalidad en paciente en diálisis, lo cierto es que la enfermedad mineral ósea sí está relacionada a eventos cardiovasculares, como lo estudia de Fukagahua et al. quienes encuentran mayor mortalidad en pacientes con hiperparatiroidismo secundario con mayor hiperfosfatemia e hipercalcemia (50).
En estudios de costos generados por hiperparatiroidismo secundario en centros de hemodiálisis de Europa se vio que las hospitalizaciones fueron mayores en pacientes con niveles de PTH por encima de 600 pg/mL. Asimismo pacientes con valores de calcio y fósforo por encima del rango normal también tuvieron incremento en el costo mensual por paciente (51).
En el estudio de Jeong et al., se encontró que la edad, los valores de prealbúmina, albúmina y el índice de masa corporal se asociaron al grado de densidad mineral ósea, no así los marcadores de recambio óseo como la fosfatasa alcalina ósea, telopéptido C y vitamina D3 en pacientes en diálisis peritoneal, lo cual nos hablaría de la importancia del aspecto nutricional en estos pacientes (52).
Se encuentra que variaciones de PTH están relacionadas con mayor muerte en pacientes, tanto en diálisis peritoneal como en hemodiálisis. Adragao et al. muestran que en pacientes en diálisis peritoneal cursan con valores menores de densidad mineral ósea detectado por DXA en el cuello femoral, ello estuvo asociado con más calificaciones, rigidez y calcificaciones vasculares periféricas (53).
Existen, además, algunos biomarcadores de la formación ósea como la fracción C terminal del propéptido de colágeno o marcadores de resorción como deoxipiridinolina (DPD)y piridinolina (PYD) En el estudio de Hampson se compararon estos marcadores con la densidad mineral ósea en la columna, el cuello femoral y el antebrazo de pacientes en diálisis peritoneal y en hemodiálisis. Se encontró que los marcadores de resorción ósea estaban más elevados en pacientes en hemodiálisis y la densidad mineral ósea más disminuida en antebrazo de pacientes en hemodiálisis, comparado con los de diálisis peritoneal (54). Otros biomarcadores estudiados son la fosfatasa alcalina y la osteoprotegerina. En el estudio de Rhee se encuentra que la fosfatasa alcalina es un buen predictor de mortalidad en pacientes con diálisis peritoneal. Se encontró, además, una asociación en la que valores de PTH por debajo de 200 y por encima de 600 pg/mL, se asociaron a mayor mortalidad (55).
En pacientes en diálisis peritoneal se midieron niveles de osteoprotegerina, que interviene en la regulación del metabolismo mineral, encontrándose que se correlacionaba con inflamación y malnutrición y, además, se sugiere considerarlo como un predictor de mortalidad cardiovascular en estos pacientes (56).
En pacientes con ERC la enfermedad mineral ósea se puede presentar como enfermedad de alto recambio (cursa con niveles séricos de paratohormona elevados) o de bajo recambio (niveles de paratohormona normales o bajos), siendo las representantes de estos fenómenos: la osteítis fibrosa para el primero y la enfermedad ósea adinámica para el segundo (1).
Lo que se espera que ocurra con el deterioro de la función renal es cierto grado de hiperactividad de la paratohormona. Sobre los factores que estarían involucrados para desarrollar una enfermedad de bajo recambio, es decir una hipofunción de la paratohormona, no han sido completamente demostrados, sin embargo, ciertos factores como edad mayor, malnutrición proteica, bajos niveles de fósforo, uso de líquidos de diálisis con gran contenido de calcio, uso excesivo de quelantes de fósforo como aluminio o calcio, uso excesivo de calcitriol o la presencia de diabetes, han sido relacionados con enfermedad de bajo recambio (57-59).
Adicionalmente, es sabido que la diálisis busca la remoción de las toxinas urémicas, una de ellas: el ácido fenilacético, este último ocasiona inhibición de la proliferación, diferenciación y mineralización de los osteoblastos, ocasionando resistencia a la paratohormona. Adicionalmente, se sabe que IL-1 e IL-6 tienen efectos inhibitorios sobre PTH y dado que dichas citokinas están elevadas en pacientes en diálisis esto permitiría una relación entre los fenómenos de malnutrición-inflamación y enfermedad de bajo recambio óseo en pacientes en diálisis. De allí la importancia de la optimización en la terapia dialítica, en la mejora del estado nutricional del paciente y en el uso racional de fármacos, de lo contrario, como ha sido demostrado en varios estudios, la frecuencia de mortalidad se incrementa (60,61).
ENFERMEDAD MINERAL ÓSEA POSTRASPLANTE RENAL
En el contexto de un paciente trasplantado renal, el estado nutricional, estado de inflamación y la exposición a esteroides representan riesgos para la persistencia o empeoramiento de las alteraciones del metabolismo mineral y óseo (62).
Con el trasplante se remueve los estímulos metabólicos que llevan al hiperparatiroidismo secundario en pacientes con enfermedad renal, sin embargo, la producción excesiva de paratohormona persiste luego del trasplante llevando a hipersecreción de PTH con hipercalcemia (hiperparatiroidismo terciario) (62-65). En pacientes trasplantados renales se ha intentado reducir rápidamente las dosis de corticoides, por los múltiples efectos metabólicos que producen, reportándose reducción de los niveles de osteocalcina y deoxipiridinolina, sin incremento en la tasa de rechazos agudos de injerto. Resultados similares se encontraron en trasplantes riñón-páncreas en un periodo de seguimiento de cuatro años (66,67).
Niveles elevados de FGF-23 podrían explicar deficiencia en la acción de calcitriol y el incremento en la pérdida renal de fósforo en el periodo postrasplante temprano, por lo que se sugiere el uso cauteloso de fármacos que puedan incrementar los niveles de fósforo séricos, pues podría llevar a una mayor secreción de FGF-23 y prolongar la pérdida renal de fósforo (68,69). Hong et al. encontraron que existían niveles disminuidos de klotho, FGFR1 y receptor sensible de calcio (CasR) en glándula paratiroides hiperplasia de pacientes trasplantados, esta situación mejoraba parcialmente con el tiempo, aunque de manera incompleta, pudiendo explicar la persistencia de hiperparatiroidismo terciario luego del trasplante renal (70).
CONCLUSIONES
En el manejo renal del calcio y fósforo intervienen una serie de proteínas, hormonas y factores que contribuyen a mantener los niveles en rango de normalidad. También tiene importancia la raza, la edad del paciente, entre otros factores.
Con la pérdida de la función renal ocurren modificaciones en los niveles de FGF-23, incluso antes de la elevación de PTH, el estímulo a los osteocitos aún no se conoce con exactitud.
En el proceso calcificación vascular intervienen promotores de calcificación FGF-23, disminución de klotho y otros factores que aún no se han identificado, pero que en conjunto promueven la calcificación, favoreciendo la aparición de eventos vasculares en pacientes con ERC.
La enfermedad mineral ósea tiene dos formas de comportamiento: alto y bajo recambio. Los factores que determinan la expresión de uno de los dos tipos no se conoce con exactitud, pero el estado de inflamación malnutrición en los pacientes en diálisis y una baja eficiencia de la misma se ha relacionado con la enfermedad de bajo recambio.
En pacientes trasplantados, la enfermedad mineral ósea cursa con ciertas modificaciones conforme transcurre el tiempo, inicialmente hay un proceso de hipofosfatemia y, en ocasiones, puede desarrollarse un hiperparatiroidismo terciario.
Fuentes de financiamiento: autofinanciada.
Conflictos de interés: los autores declaran la no existencia de algún tipo de conflicto de interés.
Referencias Bibliográficas
1. National Kidney Foundation. K/ DOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. Am J Kidney Dis. 2003 Oct;42(4 Suppl 3):S1-201.
2. Imel EA, Econs MJ. Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol. 2005 Sep;16(9):2565-75.
3. ADHR Consortium. Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF-23. Nat Genet. 2000 Nov;26(3):345-8.
4. Kuro-o M. Klotho in chronic Kidney disease-what´s new? Nephrol Dial Transplant. 2009 Jun;24(6):1705-8. doi: 10.1093/ndt/gfp069.
5. Jorgetti V, dos Reis LM, Ott SM. Ethnic differences in bone and mineral metabolism in healthy people and patients with CKD. Kidney Int. 2014 Jun;85(6):1283-9. doi: 10.1038/ ki.2013.443.
6. Kovesdy CP, Quarles LD, Lott EH, Lu JL, Ma JZ, Molnar MZ, et al. Survival advantage in black versus white men with CKD: effect of estimated GFR and case mix. Am J Kidney Dis. 2013 Aug;62(2):228-35. doi: 10.1053/j.ajkd.2012.12.012.
7. Kritchevsky SB, Tooze JA, Neiberg RH, Schwartz GG, Hausman DB, Johnson MA, et al. 25-Hydroxyvitamin D, parathyroid hormone, and mortality in black and white older adults: the health ABC study. J Clin Endocrinol Metab. 2012 Nov;97(11):4156-65. doi: 10.1210/jc.2012-1551.
8. Jovanovich A, Chonchol M, Cheung AK, Kaufman JS, Greene T, Roberts WL, et al. Racial differences in Markers of Mineral Metabolism in Advanced Chronic Kidney Disease. Clin J Am Soc Nephrol. 2012 Apr;7(4):640-7. doi: 10.2215/CJN.07020711.
9. Ho LT, Sprague SM. Women and CKDmineral bone disorder. Adv Chronic Kidney Dis. 2013 Sep;20(5):423-6. doi: 10.1053/j.ackd.2013.06.007.
10. Bacchetta J, Harambat J, Cochat J, Salusky IB, Wesseling-Perry K. The consequences of chronic kidney disease on bone metabolism and growth in children. Nephrol Dial Transplant. 2012 Aug;27(8):3063-71. doi: 10.1093/ndt/gfs299.
11. Wetzsteon RJ, Kalkwarf HJ, Shults J, Zemel BS, Foster BJ, Griffin L, et al. Volumetric bone mineral density and bone structure in childhood chronic kidney disease. J Bone Miner Res. 2011 Sep;26(9):2235-44. doi: 10.1002/ jbmr.427.
12. Yamashita T, Yoshioka M, Itoh N. Identification of a Novel Fibroblast Growth Factor, FGF-23, Preferentially Expressed in the Ventrolateral Thalamic Nucleus of the Brain. Biochem Biophys Res Commun. 2000 Oct 22;277(2):494-8.
13. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, et al. FGF-23 trangenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004 Feb 6;314(2):409-14.
14. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor induced osteomalacia. Proc Natl Acad Sci U S A. 2001 May 22;98(11):6500-5.
15. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. 2011 Feb;149(2):121-30. doi: 10.1093/jb/ mvq121.
16. Inoue Y, Segawa H, Kaneko I, Yamanaka S, Kusano K, Kawakami E, et al. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J. 2005 Aug 15;390(Pt 1):325-31.
17. Jocelak P, Olszanecka-Glinianowicz M, Chudek J. Fibroblast Growth Factor23--Structure, Function and Role in Kidney Diseases. Adv Clin Exp Med. 2012 MayJun;21(3):391-401.
18. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 Is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004 Mar;19(3):429-35.
19. Segawa H, Kawakami E, Kaneko I, Kuwahata M, Ito M, Kusano K, et al. Effect of hydrolysis-resistant FGF-23- R17Q on dietary phosphate regulation of the the renal type–II Na/ Pi transporter. Pflugers Arch. 2003 Aug;446(5):585-92.
20. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997 Nov 6;390(6655):45-51.
21. Li SA, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K. Immunohistochemical localization of Klotho protein in brain, kidney and reproductive organs of mice. Cell Struct Funct. 2004 Dec;29(4):91-9.
22. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Supression of aging in mice by the hormone Klotho. Science. 2005 Sep 16;309(5742):1829-33.
23. Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006 Jul;15(4):437-41.
24. Segawa H, Yamanaka S, Ohno Y, Onitsuka A, Shiozawa K, Aranami F, et al. Correlation between hyperphosphatemia and type II Na-Pi cotransporter activity in klotho mice. Am J Physiol Renal Physiol. 2007 Feb;292(2):F769-79.
25. Imura A, Tsuji T, Murata M, Maeda R, Kubota K, Iwano A, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007 Jun 15;316(5831):1615-8.
26. Chang Q, Hoefs S, van der Kemp AW,Topala CN,Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005 Oct 21;310(5747):490-3.
27. Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008 Jul 15;105(28):9805-10. doi: 10.1073/pnas.0803223105.
28. Huang CL. Regulation of ion channels by secreted Klotho. Adv Exp Med Biol. 2012;728:100-6. doi: 10.1007/978-1-4614-0887-1_7.
29. Torres PU, Prié D, Molina-Bletry V, Beck L, Silve C, Friedlander G. Klotho: an antiaging protein involved in mineral and vitamin metabolism. Kidney Int. 2007 Apr;71(8):730-7.
30. Pereira RC, Juppner H, AzucenaSerrano CE, Yadin O, Salusky IB, Wesseling-Perry K. Patterns of FGF- 23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone. 2009 Dec;45(6):1161-8. doi: 10.1016/j.bone.2009.08.008.
31. Larrson T, Nisbeth U, Ljunggren, Jüppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003 Dec;64(6):2272-9.
32. Ben-Dov IZ, Galitzer H, LaviMoshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007 Dec;117(12):4003-8.
33. Krajisnik T, Bjorklund P, Mansell R, Ljunggren O, Akerström G, Jonsson KB, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007 Oct;195(1):125-31.
34. Komaba H, Goto S, Fujii H, Hamada Y, Kobayashi A, Shibuya K, et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010 Feb;77(3):232-8. doi: 10.1038/ki.2009.414.
35. Gutierrez OM, Jacuzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009 May 19;119(19):2545-52. doi: 10.1161/ CIRCULATIONAHA.108.844506.
36. Nasrallah MM, El-Shehaby AR, Salem MM, Osman NA, El Sheikh E, Sharaf El Din UA. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol Dial Transplant. 2010 Aug;25(8):2679-85. doi: 10.1093/ndt/gfq089.
37. Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008 Aug 7;359(6):584-92. doi: 10.1056/ NEJMoa0706130.
38. Jean G, Terrat J, Vanel T, Hurot JM, Lorriaux C, Mayor B, et al. High levels of growing fibroblast factor (FGF)- 23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant. 2009 Sep;24(9):2792-6. doi: 10.1093/ndt/gfp191.
39. Six I, Okasaki H, Gross P, Cagnard J, Boudot C, Maizel J, et al. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS One. 2014 Apr 2;9(4):e93423. doi: 10.1371/journal.pone.0093423.
40. Kitagawa M., Suyigama H, Morinaga H, Inoue T, Takiue K, Ogawa A, et al. A decreased level of serum soluble Klotho is an independent biomarker associated with arterial stiffness in patients with chronic kidney disease. PLoS One. 2013;8(2):e56695. doi: 10.1371/journal.pone.0056695.
41. Moe SM, O’Neill KD, Duan D, Ahmed S, Chen NX, Leapman SB, et al. Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int. 2002 Feb;61(2):638-47.
42. Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008 Oct 21;118(17):1748-57. doi: 10.1161/ CIRCULATIONAHA.108.783738.
43. Chen NX, O’Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res. 2008 Nov;23(11):1798-805. doi: 10.1359/ jbmr.080604.
44. Maharem D, Gomaa S, El Ghandor M, Mohamed E, Matrawy K, Zaytoun S, et al. Association of serum fetuin-A and fetuin-A gene polymorphism in relation to mineral and bone disorders in patients with chronic kidney disease. Egypt Jour Med Gent 2013; 14(4):337- 52. doi:10.1016/j.ejmhg.2013.07.003.
45. Heiss A, Eckert T, Aretz A, Richtering W, van Dorp W, Schäfer C, et al. Hierarchial role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J Biol Chem. 2008 May 23;283(21):14815-25. doi: 10.1074/ jbc.M709938200.
46. Fang Y, Ginsberg C, Seifert M, Agapova O, Sugatani T, Register TC, et al. CKD -Induced wingless/Integration 1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J Am Soc Nephrol. 2014 Aug;25(8):1760- 73. doi: 10.1681/ASN.2013080818.
47. Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007 Jun;148(6):2635-43.
48. Thambiah S, Roplekar R, Manghat P, Fogelman I, Fraser WD, Goldsmith D, et al. Circulating sclerostin and Dickkopf-1 (DKK1) in predialysis chronic kidney disease (CKD): relationship with bone density and arterial stiffness. Calcif Tissue Int. 2012 Jun;90(6):473-80. doi: 10.1007/ s00223-012-9595-4.
49. Viaenne L, Bethes G, Claes K, Meijers B, Blocki F, Brandenburg V, et al. Sclerostin: another bone-related protein related to all-cause mortality in haemodialysis? Nephrol Dial Transplant. 2013 Dec;28(12):3024-30. doi: 10.1093/ndt/gft039.
50. Fukagahua M, Kido R, Komaba H, Onishi Y, Yamaguchi T, Hasegawa T, et al. Abnormal mineral metabolism and mortality in hemodialysis patients with secondary hyperparathyroidism: evidence from marginal structural models used to adjust for timedependent confounding. Am J Kidney Dis. 2014 Jun;63(6):979-87. doi: 10.1053/j.ajkd.2013.08.011.
51. Chirolli S, Mattin C, Belozeroff V, Perrault L, Mitchell D, Gioni I. Impact of mineral and bone disorder on healthcare resource use and associated costs in the European Fresenius medical care dialysis population: a retrospective cohort study. BMC Nephrol. 2012 Oct 29;13:140. doi: 10.1186/1471-2369- 13-140.
52. Jeong JU, Lee HK, Kim YJ, Kim JS, Kang SS, Kim SB. Nutritional markers, not markers of bone turnover, are related predictors of bone mineral density in chronic peritoneal dialysis patients. Clin Nephrol. 2010 Nov;74(5):336-42.
53. AdragaoT, Branco P, Birne R, Curto JD, de Almeida E, Prata MM, et al. Bone mineral density, vascular calcifications, and arterial stiffness in peritoneal dialysis patients. Perit Dial Int. 2008 Nov-Dec;28(6):668-72.
54. Hampson G, Vaja S, Evans C, Chesters CA, Pettit R, Evans W, et al. Comparison of the humoral markers of bone turnover and bone mineral density in patients on haemodialysis and continuous ambulatory peritoneal dialysis. Nephron. 2002 May;91(1):94-102.
55. Rhee CM, Molnar MZ, Lau WL, Ravel V, Kovesdy CP, Mehrotra R, et al. Comparative mortalitypredictability using alkaline phosphate and parathyroid hormone in patients on peritoneal dialysis and hemodialysis. Perit Dial Int. 2014 Nov-Dec;34(7):732-48. doi: 10.3747/ pdi.2013.00110.
56. Koo HM, Do HM, Kim EJ, Lee MJ, Shin DH, Kim SJ, et al. Elevated osteoprotegerin is associated with inflammation, malnutrition and new onset cardiovascular events in peritoneal dialysis patients. Atherosclerosis. 2011 Dec;219(2):925-30. doi: 10.1016/j. atherosclerosis.2011.09.025.
57. Dukkipati R, Kovesdy CP, Colman S, Budoff MJ, Nissenson AR, Sprague SM, et al. Association of relatively low serum parathyroid hormone with malnutrition-inflammation complex and survival in maintenance hemodialysis patients. J Ren Nutr. 2010 Jul;20(4):243-54. doi: 10.1053/j. jrn.2009.10.006.
58. Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004 Aug;15(8):2208-18.
59. Kalantar-Zadeh K, Kuwae N, Regidor DL, Kovesdy CP, Kilpatrick RD, Shinaberger CS, et al. Survival predictability of time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int. 2006 Aug ;70(4):771- 80.
60. Yano S, Yamaguchi T, Kanazawa I, Ogawa N, Hayashi K, Yamauchi M, et al. The uraemic toxin phenylacetic acid inhibits osteoblastic proliferation and differentiation: an implication for the pathogenesis of low turnover bone in chronic renal failure. Nephrol Dial Transplant. 2007 Nov;22(11):3160-5.
61. Allison MA, Hsi S, Wassel CL, Morgan C, Ix JH, Wright CM, et al. Calcified atherosclerosis in different vascular beds and the risk of mortality. Arterioscler Thromb Vasc Biol. 2012 Jan;32(1):140-6. doi: 10.1161/ ATVBAHA.111.235234.
62. Kunzendorf U, Krämer BK, Arns W, Braun J, Grossmann J, Pietruck F, et al. Bone disease after renal transplantation. Nephrol Dial Transplant. 2008 Feb;23(2):450-8.
63. Giovale MC, Damiano G, Lombardo C, Maione C, Buscemi G, Lo Monte AI. Bone resorption in kidney transplant recipients. Transplant Proc. 2009 May;41(4):1170-4. doi: 10.1016/j. transproceed.2009.03.043.
64. Gioviale MC, Gambino G, Maione C, Romano G, Damiano G, Cocchiara G, et al. Use of monitoring intraoperative parathyroid hormone during parathyroidectomy in patients on waiting list for renal transplantation. Transplant Proc. 2007 Jul-Aug;39(6):1775-8.
65. Rojas E, Carlini RG, Clesca P, Arminio A, Suniaga O, De Elguezabal K, et al. The pathogenesis of osteodystrophy after renal transplantation as detected by early alterations in bone remodelling. Kidney Int. 2003 May;63(5):1915-23.
66. Nishioka S, Sofue T, Inui M, Nishijima Y,Moriwaki K,Hara T, et al. Mineral and bone disorder Is temporary in patients treated with early rapid corticosteroid reduction after kidney transplantation: a single-center experience. Transplant Proc. 2014;46(2):514-20. doi: 10.1016/j.transproceed.2013.11.153.
67. Pereira S, Pedroso S, Martins L, Santos P, Almeida M, Freitas C, et al. Bone mineral density after simultaneous kidney–pancreas transplantation: four years follow-up of 57 recipients. Transplant Proc. 2010 Mar;42(2):555-7. doi: 10.1016/j. transproceed.2010.01.046.
68. Sánchez Fructuoso AI, Maestro ML, Calvo N, De La Orden V, Pérez Flores I, Vidaurreta M, et al. Role of fibroblast growth factor 23 (FGF- 23) in the metabolism of phosphorus and calcium immediately after kidney transplantation. Transplant Proc. 2012 Nov;44(9):2551-4. doi: 10.1016/j. transproceed.2012.09.070.
69. Han SY, Hwang EA, Park SB, Kim HC, Kim HT. Elevated fibroblast growth factor 23 levels as a cause of early post-renal transplantation hypophosphatemia. Transplant Proc. 2012 Apr;44(3):657-60. doi: 10.1016/j.transproceed.2011.11.046.
70. Hong YA, Choi DE, Lim SW, Yang CW, Chang YK. Decreased parathyroid Klotho expression is associated with persistent hyperparathyroidism after kidney transplantation. Transplant Proc. 2013 Oct;45(8):2957-62. doi: 10.1016/j.transproceed.2013.08.060.
Correspondencia: Jeanette Bernuy.
Dirección: Calle Pablo Usandizaga 228.
Teléfono: 999298908
Correo electrónico: jeanette.bernuy.h@upch.pe
Recibido: 28-08-14
Aprobado: 04-03-15