Artículo Original
Efecto de la administración de espironolactona sobre la pérdida de podocitos y la progresión de la nefropatía diabética experimental
Effects of spironolactone administration on the podocytes loss and progression of experimental diabetic nephropathy
Cristian Aguilar1,a, Luis Rodríguez-Delfín2,3,b
1 Departamento de Anatomía Patológica, Hospital Nacional Edgardo Rebagliati Martins. Lima, Perú.
2 Laboratorio de Biología Molecular, Facultad de Biología, Universidad Nacional Pedro Ruiz Gallo. Chiclayo, Perú.
3 Laboratorio de Investigación y Diagnóstico en Genética y Biología Molecular GEN MOL. Trujillo, Perú.
a Médico cirujano; b biólogo doctor en Ciencias con mención en Genética
RESUMEN
Objetivos. Evaluar el efecto de espironolactona (SPL) sobre la pérdida de los podocitos durante la progresión de la nefropatía diabética (ND) experimental. Materiales y métodos. Aleatoriamente un grupo de ratas macho Holtzman recibieron estreptozotocina (grupo diabético) o citrato buffer (grupo control). Las ratas diabéticas fueron tratadas con SPL (50 mg/kg/día). El área glomerular y la celularidad fueron evaluadas por métodos histomorfométricos. La lesión y pérdida de podocitos fue evaluada por la expresión de desmina y Wt-1, respectivamente. La expresión génica del TGF-β1 se evaluó mediante RT-PCR. Resultados. Los niveles de glucosa, el área glomerular, la expansión mesangial y el contenido de colágeno se incrementaron significativamente en las ratas diabéticas. La administración de SPL previno estos cambios sin modificar los niveles de glucosa. La inmunotinción para Wt-1 se redujo significativamente, mientras que la inmunotinción para desmina se incrementó drásticamente en las ratas diabéticas. El tratamiento con SPL previno el incremento de expresión de desmina y la pérdida de expresión de Wt-1. Asimismo, la administración de SPL previno el incremento de la expresión del mRNA del TGF-β1 en las ratas diabéticas. Conclusiones. El tratamiento con SPL, a través de efectos glucosa independientes, atenúa la perdida de podocitos y la progresión de los cambios morfológicos de la ND. Los presentes resultados sugieren que estos efectos son mediados, al menos en parte, por la inhibición de la la expresión del mRNA del TGF-β1.
Palabras clave: Nefropatías diabéticas; Podocitos; Fibrosis; Espironolactona; Ratas (fuente: DeCS BIREME).
ABSTRACT
Objectives. Evaluate the effect of spironolactone (SPL) on the loss of podocytes during the progression of experimental diabetic nephropathy (DN). Materials and methods. A group of male Holtzman rats randomly received streptozotocin (diabetic group) or a buffer citrate (control group). Diabetic rats were treated with SPL (50 mg/kg/day). The glomerular area and the cellularity were evaluated by histomorphometric methods. The injury and loss of podocytes was assessed by desmin expression and Wt-1, respectively. The gene expression of TGF-β1 was assessed by RT-PCR. Results. Glucose levels, the glomerular area, the mesangial expansion and collagen content increased significantly in diabetic rats. The administration of SPL prevented these changes without changing glucose levels. Immunostain for Wt-1 decreased significantly while immunostain for desmin increased dramatically in diabetic rats. Treatment with SPL prevented the increase of desmin expression and the loss of Wt-1 expression. Furthermore, the administration of SPL prevented the increase of TGF-β1 mRNA expression in diabetic rats. Conclusions. Treatment with SPL, through independent glucose effects, reduces the loss of podocytes and the progression of DN morphological changes. These results suggest that these effects are mediated, at least in part, by the inhibition of TGF-β1 mRNA expression.
Key words: Diabetic nephropathies; Podocytes; Fibrosis; Spironolactone; Rats (source: MeSH NLM).
INTRODUCCIÓN
La incidencia de la nefropatía diabética (ND) ha aumentado debido al incremento de la supervivencia de los pacientes diabéticos. En muchos países, incluyendo el Perú, la ND se ha convertido en una de las causas más frecuentes de enfermedad renal terminal (1). Numerosos estudios han demostrado que la pérdida de podocitos junto con la expansión mesangial y la fibrosis túbulo-intersticial son las manifestaciones estructurales más precoces del daño glomerular y constituyen los principales determinantes del curso clínico de la ND (2-3).
Evidencia creciente sugiere un papel fundamental del podocito en el desarrollo de la proteinuria y en la progresión de la disfunción glomerular (2). A diferencia de otras células del glomérulo, los podocitos tienen una capacidad limitada para replicarse, por lo tanto, su pérdida conduce inevitablemente al desarrollo de glomeruloesclerosis (2,3). De hecho, estudios morfométricos en pacientes con diabetes de corta duración muestran una progresiva depleción de podocitos (4). Asimismo, se ha demostrado que en la diabetes se encuentra incrementada la eliminación urinaria de podocitos desde estadios tempranos (5). Estos hallazgos permiten sugerir que la pérdida de podocitos precede al desarrollo de glomeruloesclerosis en la ND.
La videncia clínica y experimental sugiere que la activación y la redistribución de los componentes del sistema renina-angiotensina-aldosterona (SRAA) y su principal efector, la angiotensina II (Ang II), cumplen una función central en el remodelado estructural del riñón diabético y su progresión a la glomeruloesclerosis (2,6). Durante mucho tiempo se ha creído que la inhibición del SRAA con inhibidores de la enzima de conversión de angiotensina (IECA) bloquea la producción de aldosterona. Sin embargo, se ha descrito que el bloqueo del SRAA con IECA no produce necesariamente un descenso mantenido de los niveles de aldosterona, sino que esta aumenta progresivamente a lo largo del tiempo (fenómeno conocido como “escape de aldosterona”) (6). Asimismo, diversos estudios clínicos han demostrado que la espironolactona (SPL), un bloqueador de la aldosterona, mejora la proteinuria de los pacientes con ND, aun en aquellos donde el tratamiento con IECA no ha sido efectivo (7).
La etiología exacta de la pérdida de podocitos en la diabetes sigue siendo especulativa. Se considera que tanto la apoptosis como el desprendimiento celular inducido por el factor de crecimiento transformante β1 (TGF-β1cumplen un rol central en este proceso (8). De hecho, nefropatías de diferente etiología, así como la ND, se caracterizan por presentar un marcado incremento en la expresión del mRNA del TGF-β1 (9). Asimismo, se ha demostrado que los componentes del SRAA, incluyendo la aldosterona, son capaces de activar la síntesis del TGF-β1 en el riñón diabético (7,8) y que la infusión de aldosterona a ratas normales incrementa la eliminación urinaria del TGF-β1, incluso, sin modificar los niveles de presión arterial (10). De la misma forma, se ha demostrado que la administración de IECA previene la sobreexpresión del TGF-β1 en los podocitos y atenúa la progresión de la glomeruloesclerosis (11). Evidencia reciente sugiere que el bloqueo selectivo de la aldosterona tiene un efecto beneficioso sobre la progresión de la disfunción renal (12-14), no solamente a través de sus efectos hemodinámicos, sino también a través de su capacidad para regular tanto la expresión basal como la expresión inducida del TGF-β1 en células renales (15-17).
La pérdida de podocitos conduce a glomeruloesclerosis progresiva y, en último término, al deterioro de la función renal. Shibata et al. (18) mostraron que la infusión de aldosterona en ratas provoca la pérdida de podocitos y que el bloqueo del receptor mineralocorticoide previene dicho fenómeno. Sin embargo, a la fecha, el efecto del bloqueo de la aldosterona sobre la pérdida de podocitos en una etapa temprana de la ND sigue siendo incierto. Por consiguiente, el objetivo de este estudio fue determinar el efecto de la administración de SPL sobre la pérdida de podocitos y los cambios estructurales asociados con la progresión de la ND en ratas diabéticas.
MATERIALES Y MÉTODOS
El protocolo experimental de este estudio se realizó de acuerdo con la Guía para el cuidado y uso de animales de laboratorio, publicado por el National Institutes of Health (NIH Publication, 1996). La diabetes fue inducida en ratas macho Holtzman (200-220 g) con una inyección intraperitoneal de estreptozotocina (STZ) 50 mg/kg preparado en 0,1 mol/L de buffer citrato (pH 4,5). Solo las ratas con niveles de glucosa sérica mayor a 200 mg/dL tres días después de la inyección de STZ fueron incluidas en el estudio. Las ratas control fueron inyectadas con el mismo volumen de buffer citrato.
PROTOCOLO Y GRUPOS EXPERIMENTALES
Los animales se distribuyeron aleatoriamente en tres grupos experimentales: (i) ratas control sin diabetes (n=8); (ii) ratas diabéticas sin tratamiento (STZ, n=8) y (iii) ratas diabéticas tratadas con espironolactona (STZ-SPL, n=8). La SPL se administró en dosis de 50 mg/kg/ día por gavaje diario. El tratamiento se continúo durante doce semanas. Los animales fueron mantenidos bajo condiciones estándar de iluminación (ciclo luz-oscuridad de doce horas) y alimentados con una dieta estándar y agua ad libitum. Al final del seguimiento, se procedió a pesar y sacrificar a los animales tras haber obtenido muestras de sangre para las determinaciones bioquímicas. Inmediatamente después del sacrificio se practicó una laparotomía mediana para extraer ambos riñones.
Al final del seguimiento, las ratas fueron sometidas a un ayuno de 12 h, luego del cual se tomaron muestras de sangre por punción cardiaca. El suero fue separado y posteriormente se analizó los niveles de glucosa y creatinina por colorimetría (biochemical diagnostic kit, Human, Germany) mediante un espectrofotómetro UV/Vis (Jenway 6505 UV/VIS, Felsted, UK).
DETERMINACIÓN DEL REMODELADO RENAL POR HISTOMORFOMETRÍA
La relación entre el peso renal (PR) y el peso corporal de la rata (PC) fue utilizada para estimar la masa renal relativa (PR/PC). Posteriormente, el tejido renal fue fijado en formalina, embebido en parafina y coloreado con la tinción ácido periódica de Schiff (PAS) y tricromica de Masson. Para estimar la hipertrofia glomerular, el área de cada glomérulo (Ag) se determinó trazando manualmente la superficie de la cápsula de Bowman con la ayuda de un planímetro digital (ImageJ, NIH, USA) en 25 glomérulos de cada espécimen. El índice de expansión mesangial (IEM) se estimó a través del cálculo del porcentaje del área glomerular ocupada por el mesangio en 25 glomérulos consecutivos de cada muestra coloreada con PAS (16). Un solo investigador (CA) desconocedor de la naturaleza de los grupos experimentales estuvo a cargo del análisis histológico.
ANÁLISIS INMUNOHISTOQUÍMICO DE LA CORTEZA RENAL
La inmunotinción fue realizada aplicando el método estreptavidina-biotina peroxidasa con ligeras modificaciones. Brevemente los tejidos fijados fueron desparafinados y rehidratados. Luego de la recuperación antigénica, los cortes se incubaron con el anticuerpo monoclonal contra los antígenos del gen supresor del tumor de Wilms (Wt-1; 1:100; Dako, CA, USA) y Desmina (Desmin; 1:100; Dako, CA, USA). La inmunotinción se reveló con el cromógeno DAB (3, 3´- diaminobenzidine) y las secciones fueron contrastadas con hematoxilina. El número de podocitos por glomérulo se determinó por el número de células que mostraban expresión nuclear para Wt-1 dentro del glomérulo. La intensidad de expresión de desmina en el glomérulo se determinó en base al porcentaje del área glomerular ocupado por las células inmunoreactivas.
DETERMINACIÓN DE LA EXPRESIÓN GÉNICA EN LA CORTEZA RENAL
Para determinar si la transcripción del TGF-β1 en la corteza renal se vio afectada por la diabetes, la expresión del mRNA del TGF-β1 se evaluó mediante transcripción reversa acoplada a la reacción en cadena de ADN polimerasa (RT-PCR). El ARN total fue extraído de la corteza renal utilizando el kit PureLink™ Micro-to-Midi Total RNA Purification System (Invitrogen, CA, USA). Diez microlitros de ARN total fue utilizado para sintetizar la cDNA usando el kit SuperScript III First-Strand Synthesis (Invitrogen, CA, USA). El cDNA fue amplificado por PCR mediante el kit Platinum®Taq DNA Polymerase (Invitrogen, CA, USA) para TGF-β1 y gliceraldehído-3-fosfato deshidrogenasa (GAPDH), utilizando las siguientes secuencias de primers: TGF-β1 sentido 5’-ACCGCAACAACGCAATCTAT-3’; TGF-β1 antisentido 5’-ACGCCAGGAATTGTTGCTAT-3’; GAPDH sentido 5’-CCCTCAAGATTGTCAGCAATGC-3’; GAPDH antisentido 5’-GTCCTCAGTGTAGCCCAGGAT-3’ (16), 30 ciclos fueron usados para amplificar cada gen. La intensidad de las bandas se cuantificó por densitometría (ImageJ, NIH, USA) en gel de poliacrilamida al 4% y se normalizó con respecto a la banda del gen control GADPH, con expresión estándar en la corteza renal.
DETERMINACIÓN DEL CONTENIDO DE COLÁGENO
La determinación del contenido de colágeno renal se realizó mediante un método bioquímico basado en la cuantificación de hidroxiprolina (HOP) modificado (16). Brevemente, el tejido (50 mg) fue homogeneizado manualmente en agua destilada. Los homogeneizados se hidrolizaron en un volumen de NaOH 4N a 120 °C durante 20 min. Las muestras se oxidaron con 450 μL de reactivo de cloramina T durante 25 min a temperatura ambiente. Las muestras se mezclaron luego con 500 μL de reactivo de Erlich e incubados a 65 °C durante 20 min. La absorbancia de la muestra fue leída a 550 nm en un espectrofotómetro (Jenway 6505 UV/VIS, Felsted, UK). El contenido de colágeno fue estimado a partir de una curva de calibración (0-10 μg/mL de la L-hidroxiprolina), suponiendo que el contenido de HOP representa el 12,5% del tejido colágeno (19).
ANÁLISIS ESTADÍSTICO
Los resultados fueron expresados como medianas y rangos intercuartílicos. Se aplicó la prueba de Kruskal Wallis para establecer la diferencia entre los grupos, con un nivel de confianza al 95%. Seguido de una prueba de Wilcoxon para identificar las diferencias específicas entre grupos. Los datos fueron procesados con el paquete estadístico STATA 12.1.
RESULTADOS
EFECTO SOBRE LOS PARÁMETROS METABÓLICOS
Al final del seguimiento, la exposición a STZ se asoció con un incremento en la glucosa sérica de los animales diabéticos en relación al grupo control (409,1 [14,9] mg/dL frente a 116,1 [14,9 ] mg/dL, p<0,001). El tratamiento de las ratas diabéticas con SPL no modificó los niveles de glucosa (406,9 [30,4] mg/dL). Con relación a los niveles de creatinina sérica no se encontraron cambios significativos entre los tres grupos experimentales (p=0,44).
EFECTO SOBRE LA HISTOMORFOMETRÍA RENAL
La relación PR/PC se incrementó significativamente en el grupo STZ (1,04 [0,14]) con respecto al grupo control (0,79 [0,08]; p<0,001). El incremento de la relación PR/PC fue similar en las ratas del grupo STZ-SPL en comparación con las ratas del grupo STZ (0,98 [0,17] frente a 1,04 [0,14]; p=0,32). A la microscopia óptica, la hipertrofia glomerular es el cambio estructural más precoz de la ND. El área glomerular (Ag) se incrementó significativamente en el grupo diabético (10 240 [0,49] μm2) comparado con el grupo no diabético (7 950 [0,47] μm2; p=0.001). En el grupo STZ-SPL el Ag fue significativamente menor (8 250 [0,44] μm2) con respecto al grupo STZ (10 240 [0,49] μm2; p=0.001) (Tabla 1).
EFECTO SOBRE LA EXPANSIÓN MESANGIAL INDUCIDA POR STZ
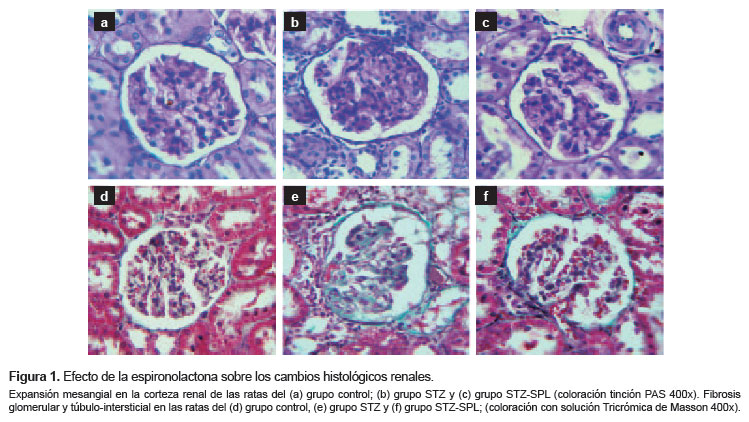
Las ratas diabéticas aumentaron en 2,5 veces la acumulación de matriz PAS-positiva (25,5 ± [5,10]) en comparación con el grupo control (9,8 [3,46]; p=0,001). Este parámetro histológico fue significativamente menor en el grupo STZ-SPL, en relación al grupo STZ (15,4 [3,58] frente a 25,5 [5,10]; p=0,001). Estas observaciones morfológicas sugieren una acumulación significativa de proteína PAS-positiva en la matriz mesangial de las ratas diabéticas no tratadas. Asimismo, el análisis histológico sugiere que la hiperglucemia provoca un incremento del depósito de matriz extracelular (MEC) en el mesangio, hiperplasia de las células mesangiales, y adhesión de la cápsula glomerular en las ratas diabéticas. El tratamiento con SPL logró reducir estas alteraciones (Figura 1a, 1b, 1c).
EFECTO SOBE EL INCREMENTO DEL CONTENIDO DE HIDROXIPROLINA RENAL
La hiperglucemia incrementó en 1,6 veces el contenido de HOP del riñón de las ratas diabéticas en comparación con las ratas control (3,77 [1,20] μg/mg frente a 2,10 [0,54] μg/mg; p=0,001). En el grupo STZ-SPL el contenido de HOP renal fue significativamente menor en relación a las ratas del grupo STZ (2,75 [0,71] μg/mg frente a 3,77 [1,20] μg/mg; p<0,02). Del mismo modo, el análisis histológico sugiere un incremento marcado del depósito de MEC en los riñones de las ratas diabéticas principalmente en la matriz mesangial glomerular y la membrana basal de los túbulos renales. En el grupo STZ-SPL existe una atenuación parcial de estas alteraciones (Figura 1d, 1e, 1f).
EFECTO SOBE LA PÉRDIDA DE PODOCITOS
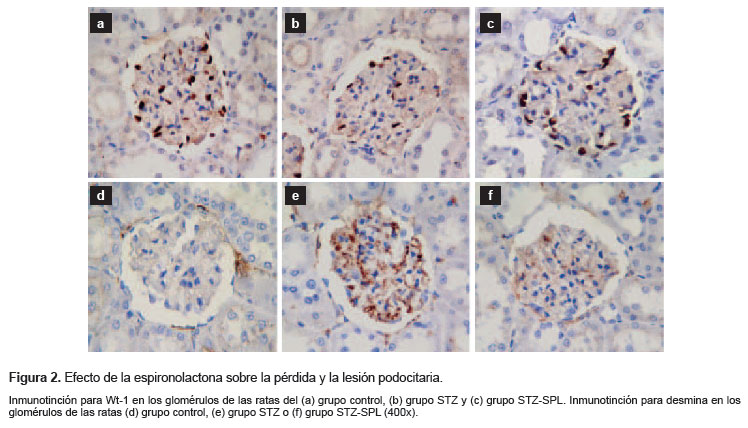
Para evaluar la pérdida de podocitos se cuantificó el número de células que mantenían expresión nuclear de Wt-1 en cada glomérulo. En comparación con las ratas control, el análisis de células con núcleo Wt-1 positivo por glomérulo mostró una reducción significativa en el grupo STZ (10 [2,5] frente a 17,5 [4,5]; p<0,001). El grupo STZ-SPL tuvo una menor pérdida de podocitos en comparación las ratas STZ (15 [2] frente a 10 [2,5]; p<0,001) (Figura 2a, 2b, 2c).
EFECTO SOBRE LA LESIÓN PODOCITARIA
La inmunotinción para desmina, un marcador precoz de lesión podocitaria, estuvo incrementada en el glomérulo de ratas del grupo STZ en comparación con las ratas control (2,25 [1] frente a 0,62 [1]; p=0,001). El grupo STZ-SPL tuvo una menor expresión de desmina en el glomérulo en comparación con el grupo STZ (1,12 [1,5] frente a 2,25 [1]; p=0,017) (Figura 2d, 2e, 2f).
EFECTO SOBRE LA EXPRESIÓN DEL mRNA TGF-β1
El remodelado renal inducido por la hiperglicemia se asocia con un incremento en la expresión del TGF-β1. Estos resultados muestran un incremento significativo en 2,4 veces los niveles de mRNA para TGF-β1 en la corteza renal de las ratas del grupo STZ respecto a las ratas control (1,41 [0,20 frente a 0,58 [0,24]; p=0,001). En el grupo STZ-SPL la expresión génica para TGF-β1 fue significativamente menor respecto al grupo STZ (0,79 [0,42] frente a 1,41 [0,20]; p=0,01).
DISCUSIÓN
La pérdida de podocitos en una etapa subclinica ha sido recientemente reconocida como un importante mediador en la progresión del daño glomerular (2,4). Los podocitos son células epiteliales altamente diferenciadas que cumplen un papel fundamental en el mantenimiento de la estructura y función de la barrera de filtración glomerular, por lo tanto, la pérdida de estas células condiciona el desarrollo progresivo de disfunción renal. Diversos estudios clínicos y experimentales de daño glomerular han demostrado que la reducción progresiva en el número de podocitos precede al desarrollo de glomeruloesclerosis (2,4,5,8). Debido a su limitada capacidad proliferativa, el desprendimiento de podocitos de la membrana basal glomerular (MBG) conduce a la depleción celular. En este contexto, se cree que la MBG desnuda se adhiere a la cápsula de Bowman y favorece la formación de sinequias, iniciando el desarrollo de glomeruloesclerosis (8).
Bajo condiciones fisiológicas, solamente las células mesangiales muestran una tenue expresión de desmina a nivel glomerular. Sin embargo, durante la progresión de diversos modelos de nefropatía se ha demostrado que el daño renal condiciona una marcada expresión de desmina en los podocitos lesionados (18). Recientemente se ha reportado la capacidad de SPL para prevenir la expresión de desmina en podocitos lesionados en un modelo animal de nefropatía (20). Nuestros hallazgos muestran que la administración de SPL logró prevenir la sobrexpresión de desmina en el glomérulo de ratas diabéticas (Figura 2). Los podocitos maduros bien diferenciados se caracterizan por expresar Wt-1 continuamente, por lo tanto la reducción en la densidad de los podocitos que expresan Wt-1 refleja un intenso daño podocitario e incluso puede ser considerado un marcador precoz de su apoptosis (21,22). En el presente estudio el tratamiento con SPL previno la pérdida de podocitos en las ratas diabéticas (Figura 2). Del mismo modo, Toyonaga et al. (23) han demostrado que SPL es capaz de prevenir la pérdida de expresión de Wt-1 en podocitos inducida por la diabetes mediante la reducción del estrés oxidativo.
Estudios clínicos han demostrado que el aldosteronismo primario se asocia con una albuminuria excesiva comparada con la producida por la hipertensión esencial (18). Asimismo, se ha demostrado que el bloqueo de aldosterona reduce la albuminuria en los pacientes hipertensos. Estas observaciones sugieren que la aldosterona puede causar directamente la disfunción de la barrera de filtración glomerular. Aunque los mecanismos exactos de lesión renal inducida por la aldosterona aún no están claros, la evidencia acumulada indica que la aldosterona tiene efectos directos sobre diversas células renales, incluyendo los podocitos, y que puede modular sus funciones a través de acciones profibróticas, del estrés oxidativo, y la alteración de los reguladores del ciclo celular (8,18). Además, interesa observar que los podocitos expresan receptores mineralocorticoides y que la activación de dichos receptores disminuye la expresión de proteínas integrales de su diafragma de filtración, como nefrina y podocina, favoreciendo su desprendimiento (8). Más allá de las alteraciones funcionales, se ha demostrado que la aldosterona también es capaz de alterar la estructura del cuerpo y los pedicelos de los podocitos. Asimismo, resulta sorprendente observar que estas alteraciones se pueden atenuar cuando se administran inhibidores de la aldosterona (18).
Dentro de las lesiones glomerulares observadas durante la progresión de la ND, la expansión de la matriz mesangial es una de las más frecuentes (2,3). Este tipo de daño glomerular puede llevar a la reducción de la superficie de filtración glomerular, condicionando así una seria limitación para la ultrafiltración glomerular. De hecho, existe una relación directa entre la expansión de la matriz mesangial y la pérdida de la función renal en los portadores de ND (3). Yuan et al. (24) han demostrado que el bloqueo selectivo de aldosterona mediante SPL es capaz de prevenir el aumento de la expansión mesangial en un modelo de ND. Nuestros resultados muestran que el incremento de la expansión mesangial en las ratas diabéticas fue reducido significativamente por la administración de SPL (Figura 1). Por otro lado, es ampliamente conocido que la hiperglicemia per se, además de incrementar el depósito de MEC, es capaz de inducir alteraciones funcionales en la célula mesangial que inducen la sobreexpresión de diversos factores de crecimiento que favorecen el desarrollo precoz de hipertrofia glomerular (2). Nuestros hallazgos demuestran que la administración de SPL previene el incremento del área glomerular en las ratas diabéticas (Tabla 1). De la misma forma, Pereira et al. (25), demostraron en un modelo de ND que SPL era capaz de prevenir el desarrollo de hipertrofia glomerular, sin modificar los niveles de presión arterial.
El grado de lesión túbulo-intersticial, en especial la extensión de la fibrosis intersticial, se correlaciona estrechamente con la función renal en una variedad de enfermedades glomerulares primarias (26). En un contexto de hiperglucemia, TGF-β1 constituye el principal factor condicionante para el desarrollo de fibrosis renal, al estimular el incremento de MEC y la transición epitelio-mesenquimal (TEM) en el intersticio renal (8). Estudios recientes han demostrado que el incremento de aldosterona intrarrenal se asocia con un marcado incremento de la síntesis de mRNA del TGF-β1 (27) y que el bloqueo selectivo de aldosterona reduce la síntesis de colágeno en el riñón (15,16). Fujisawa et al. (17) fueron los primeros en reportar la capacidad de SPL para atenuar la fibrosis renal a través de la inhibición de la expresión del mRNA del TGF-β1 en ratas diabéticas. Nuestros resultados muestran que la administración de SPL previene significativamente el depósito del hidroxiprolina y la extensión de fibrosis intersticial en el riñón diabético, efecto que sería mediado, al menos en parte, por la inhibición de la sobreexpresión del mRNA del TGF-β1 inducida por la diabetes.
La disfunción del podocito y la producción local del TGF-β1 han sido fuertemente implicadas en la patogénesis de la glomeruloesclerosis (8,9,11). De hecho, a pesar del carácter multifactorial de la esclerosis glomerular, numerosos estudios han identificado colectivamente la pérdida del podocito como un evento central en este fenómeno (2,4,8,9,11). Asimismo, se ha demostrado la capacidad de los podocitos para sintetizar TGF-β1 en respuesta a las concentraciones elevadas de glucosa (11). Resulta interesante apreciar que a diferencia de las células mesangiales, los podocitos no sobreexpresan TGF- β1, cuando se exponen a Ang II (28). Del mismo modo, se ha reportado que el incremento de la síntesis del TGF-β1 en los podocitos induce la transformación fenotípica de las células mesangiales, favoreciendo el inicio de la glomeruloesclerosis (8,11). Aunque, el mecanismo subyacente por el cual aldosterona induce lesión renal (en general) y en los podocitos (en particular) no es bien comprendido, se sugiere que el incremento de la expresión del TGF-β1 inducido por la aldosterona tendría un rol importante en este proceso (8,9,11,18,26). De hecho, más allá del incremento de la síntesis de MEC, el TGF-β1 tiene efectos directos sobre la proliferación, hipertrofia y apoptosis en las células renales (8).
Durante la década pasada la Ang II ha sido propuesta como el principal mediador del remodelado renal que precede a la insuficiencia renal, independientemente del daño inicial. Sin embargo, recientemente diversos reportes experimentales han señalado la capacidad de la aldosterona para contribuir directamente a la progresión del remodelado estructural en diversos modelos de nefropatía, incluso a través de efectos independientes a la generación de Ang II (29,30). Del mismo modo, numerosos ensayos clínicos han reportado que el bloqueo de la aldosterona tiene efectos beneficiosos independientes al bloqueo del SRAA en la ND (12-14). Nuestros hallazgos nos permiten sugerir que los efectos nefroprotectores de SPL en el riñón diabético estarían mediados, al menos en parte, por la inhibición de la sobreexpresión del mRNA del TGF-β1 inducida por aldosterona.
En la presente investigación, la administración de SPL no afectó los niveles séricos de glucosa en las ratas diabéticas tratadas. Esto nos permite corroborar que SPL no ejerció efecto metabólico significativo en el control glucémico de las ratas diabéticas. Esta observación nos permite sugerir que SPL reduce la pérdida de podocitos, la proliferación celular y la síntesis de colágeno, independientemente de su efecto sobre la glucosa.
Concluimos que el remodelado renal observado en la ND se acompaña de una sobreexpresión del TGF-β1, sugiriendo su participación en la pérdida de podocitos y el depósito exagerado de MEC. La administración de SPL no solamente inhibió la sobreexpresión del TGF-β1 sino que también atenúo la perdida de podocitos y los cambios morfológicos del remodelado renal, a través de efectos glucosa independientes; sugiriendo un rol potencial de este fármaco en el manejo de la ND.
Contribuciones de autoría: CA participó en la concepción y diseño del trabajo, análisis e interpretación de datos, obtención de financiamiento y redacción del manuscrito. LRD participó en el diseño del estiudio y las pruebas de RT-PCR, interpretación y redacción parcial del manuscrito. Ambos autores revisaron y aprobaron la versión final del trabajo.
Fuentes de financiamiento: autofinanciada.
Conflictos de interés: los autores declaran no tener conflictos de interés en la publicación de este artículo.
Referencias Bibliográficas
1. Hurtado A, Escudero E, Stromquist CS, Urcia J, Hurtado ME, Gretch D, et al. Distinct patterns of glomerular disease in Lima, Peru. Clin Nephrol. 2000;53(5):325-32.
2. Kriz W, Lemley KV. The role of the podocyte in glomerulosclerosis. Curr Opin Nephrol Hypertens. 1999;8(4):489-97.
3. Steffes MW, Osterby R, Chavers B, Mauer SM. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes. 1989;38(9):1077-81.
4. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99(2):342-8.
5. Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Ebihara I, et al. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15(9):1379-83.
6. Athyros VG, Mikhailidis DP, Kakafika AI, Tziomalos K, Karagiannis A. Angiotensin II reactivation and aldosterone escape phenomena in renin-angiotensin-aldosterone system blockade: is oral renin inhibition the solution? Expert Opin Pharmacother. 2007;8(5):529-35.
7. Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of Angiotensin Receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2641-50.
8. Lee HS. Pathogenic role of TGF-β in the progression of podocyte diseases. Histol Histopathol. 2011;26(1):107-16.
9. Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, et al. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different satges of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia. 2005;48(12):2650-60.
10. Juknevicius I, Segal Y, Kren S, Lee R, Hostetter TH. Effect of aldosterone on renal transforming growth factor-beta. Am J Physiol Renal Physiol. 2004;286(6):F1059-62.
11. Abbate M, Zoja C, Morigi M, Rottoli D, Angioletti S, Tomasoni S, et al. Transforming growth factor-beta1 is up-regulated by podocytes in response to excess intraglomerular passage of proteins: a central pathway in progressive glomerulosclerosis. Am J Pathol. 2002;161(6):2179-93.
12. Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006;70(12):2116-23.
13. Chrysostomou A, Pedagogos E, MacGregor L, Becker GJ. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin II receptor blocker. Clin J Am Soc Nephrol. 2006;1(2):256-62.
14. Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005;28(9):2106-12.
15. Guney I, Selcuk NY, Altintepe L, Atalay H, Başarali MK, Büyükbaş S. Antifibrotic effects of aldosterone receptor blocker (spironolactone) in patients with chronic kidney disease. Ren Fail. 2009;31(9):779-84.
16. Taira M, Toba H, Murakami M, Iga I, Serizawa R, Murata S, et al. Spironolactone exhibits direct renoprotective effects and inhibits renal renin-angiotensin-aldosterone system in diabetic rats. Eur J Pharmacol. 2008;589(1-3):264-71.
17. Fujisawa G, Okada K, Muto S, Fujita N, Itabashi N, Kusano E, et al. Spironolactone prevents early renal injury in streptozotocin-induced diabetic rats. Kidney Int. 2004;66(4):1493-502.
18. Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocytes as target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49(2):355-64.
19. Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem. 1996;29(3):225-9.
20. Nishiyama A, Kobori H, Konishi Y, Morikawa T, Maeda I, Okumura M, et al. Mineralocorticoid receptor blockade enhances the antiproteinuric effect of an angiotensin II blocker through inhibiting podocyte injury in type 2 diabetic rats. J Pharmacol Exp Ther. 2010;332(3):1072-80.
21. Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D, Hammes A, et al. WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet. 2002;11(6):651-9.
22. Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225-33.
23. Toyonaga J, Tsuruya K, Ikeda H, Noguchi H, Yotsueda H, Fujisaki K, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant. 2011;26(8):2475-84.
24. Yuan J, Jia R, Bao Y. Beneficial effects of spironolactone on glomerular injury in streptozotocin-induced diabetic rats. J Renin Angiotensin Aldosterone Syst. 2007;8(3):118-26.
25. Pereira LM, Querido DL, Madeira AC, Mandarim-De-Lacerda CA. Volume-weighted mean glomerular volume in spontaneously hypertensive rats treated with different doses of spironolactone. Int Braz J Urol. 2002;28(4):356-62..
26. Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999;56(5):1627-37.
27. Han JS, Choi BS, Yang CW, Kim YS. Aldosterone-induced TGF-beta1 expression is regulated by mitogen-activated protein kinases and activator protein-1 in mesangial cells. J Korean Med Sci. 2009;24 Suppl:S195-203.
28. Chen S, Lee JS, Iglesias-de la Cruz MC, Wang A, Izquierdo-Lahuerta A, Gandhi NK, et al. Angiotensin II stimulates alpha3(IV) collagen production in mouse podocytes via TGF-beta and VEGF signalling: implications for diabetic glomerulopathy. Nephrol Dial Transplant. 2005;20(7):1320-8.
29. Aldigier JC, Kanjanbuch T, Ma LJ, Brown NJ, Fogo AB. Regression of existing glomerulosclerosis by inhibition of aldosterone. J Am Soc Nephrol. 2005;16(11):3306-14.
30. Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest. 1996;98(4):1063-8
Correspondencia: Luis Rodríguez Delfín
Dirección: Laboratorio de Biología Molecular Facultad de Ciencias Biológicas Medicina, Universidad Nacional de Pedro Ruiz Gallo. Lambayeque, Perú.
Teléfono: (511) 44 310210
Correo electrónico: ladelfin@amauta.rcp.net.pe
Recibido: 24-08-12
Aprobado: 14-11-12