
ORIGINAL BREVE
Recuento poblacional linfocitario como primera aproximación al diagnóstico de inmunodeficiencias primarias
Lymphocyte population count as a first approach to the diagnosis of primary immunodeficiencies
David García-Gomero1,2,5,a, Edgar Matos-Benavides1,3,5,b, Rosario Inocente-Malpartida1,2,5,c, Daniel Mendoza-Quispe3,a, Joel Chalco-Huamán3,a, María Lopez-Talledo1,5,b, Wilmer Córdova-Calderón1,5,b, Raúl Muñoz-León1,5,b, César Galván-Calle1,5,b
1 Centro Nacional de Referencia de Alergia, Asma e Inmunología del Instituto Nacional de Salud del Niño-Breña. Lima, Perú.
2 Facultad de Medicina, Escuela de Posgrado, Universidad Peruana Cayetano Heredia (UPCH). Lima, Perú.
3 Facultad de Medicina, Universidad Nacional Mayor de San Marcos (UNMSM). Lima, Perú.
4 Facultad de Ciencias Biológicas, Escuela de Posgrado. Universidad Nacional Mayor de San Marcos (UNMSM). Lima, Perú.
5 Sociedad Peruana de Inmunología. Lima, Perú.
a Médico cirujano; b médico inmunólogo clínico-alergólogo; c tecnólogo médico.
RESUMEN
Las inmunodeficiencias primarias (IDP) se caracterizan por alteraciones de los componentes del sistema inmunitario. El recuento poblacional linfocitario por citometría de flujo es una aproximación al diagnóstico molecular y se expresa por inmunofenotipos. El objetivo del estudio fue describir el recuento poblacional linfocitario y los inmunofenotipos compatibles con IDP en pacientes con sospecha de IDP en un hospital de referencia nacional peruano. Se revisaron los registros de 261 casos que cumplían con los criterios de sospecha clínica para IDP de la Jeffrey Modell Foundation entre abril y diciembre de 2016. De los 261 casos con sospecha de IDP se hallaron 54,8% de varones. Se encontró 93 pacientes (35,6%) con inmunofenotipos compatibles con alguna IDP. El inmunofenotipo de inmunodeficiencia común variable fue más frecuente (36,6%), seguido de agammaglobulinemias (18,3%). Las deficiencias de anticuerpos fueron las IDP más frecuentes. Es necesario realizar otras pruebas moleculares para el diagnóstico genético específico.
Palabras clave: Síndromes de Inmunodeficiencia; Citometría de Flujo; Diagnóstico (fuente: DeCS BIREME).
ABSTRACT
Primary immunodeficiencies (PID) are characterized by alterations in the components of the immune system. The lymphocyte population count by flow cytometry is an approach to molecular diagnosis and is expressed by immunophenotypes. The objective of the study was to describe the lymphocyte population count and immunophenotyping compatible with PID in patients with suspected PID in a Peruvian national reference hospital. Records of 261 cases meeting the Jeffrey Modell Foundation’sPID clinical suspicion criteria were reviewed between April and December of 2016. Of the 261 suspected cases of PID, 54.8% were males. We found 93 patients (35.6%) with PID-compatible immunophenotyping. The common variable immunodeficiency immunophenotype was the most frequent (36.6%), followed by agammaglobulinemias (18.3%). Antibody deficiencies were the most common PID. Other molecular tests are needed for a specific genetic diagnosis.
Keywords: Immunological Deficiency Syndromes; Flow Cytometry; Diagnosis (source: MeSH NLM).
INTRODUCCIÓN
Las inmunodeficiencias primarias (IDP) son un conjunto de enfermedades con trasfondo genético que se derivan en alteraciones del número y/o función de los elementos del sistema inmunitario (1). Su prevalencia a nivel global se estima en uno por cada 2000 nacidos vivos (2). En el contexto peruano, se ha reportado un total de 176 casos a un registro internacional para el 2017, aunque se esperaría un total de 300 casos diagnosticados al año, según la prevalencia global y la expectativa de nacidos vivos en el Perú (3). Este infradiagnóstico, se derivaría de la falta de equipos de diagnóstico a nivel de los países latinoamericanos para realizar pruebas moleculares (4), tales como, la citometría de flujo, análisis T REC/K REC, secuenciamiento genético, pruebas vacunales, etc.
La citometría de flujo, entre sus múltiples aplicaciones, permite realizar un recuento de subpoblaciones de linfocitos gracias a la detección inmunológica de marcadores de linfocitos T (TCD3+, TCD4+, TCD8+ y NK CD16/56+) y de linfocitos B (CD19+) (5). Así, el recuento poblacional linfocitario permitiría establecer inmunofenotipos compatibles con las inmunodeficiencias primarias más frecuentes: agammaglobulinemias con linfocitos B CD19+ ausentes, inmunodeficiencia común variable con linfocitos B CD19+ disminuidos, inmunodeficiencias combinadas severas con linfocitos TCD3+ disminuidos y posibles alteraciones a nivel de linfocitos B CD19+, y linfocitos T NK CD16/56+(6).Se ha evidenciado que los inmunofenotipos compatibles con inmunodeficiencia común variable (ICV) y con agammaglobulinemias son los más frecuentes (7,8). El hallazgo de estos inmunofenotipos en relación con los datos clínicos previamente obtenidos permitiría aproximarnos al diagnóstico de IDP y ser útiles para indicar pruebas específicas de diagnóstico genético por secuenciamiento (9).
El Centro de Referencia Nacional de Alergia, Asma e Inmunología (CERNAAI) del Instituto Nacional de Salud del Niño (INSN) implementó el primer laboratorio público de inmunodiagnóstico donde ha sido posible realizar pruebas moleculares como la citometría de flujo. Bajo ese contexto, se realizó un estudio cuyo objetivo fue describir el recuento poblacional linfocitario y los inmunofenotipos compatibles con IDP en pacientes con sospecha de IDP en un hospital de referencia nacional del Perú.
EL ESTUDIO
El presente estudio fue descriptivo y consistió en revisión de historias clínicas y registros de todas las citometrías de flujo para descarte de IDP realizadas entre abril y diciembre del 2016 en el CERNAAI del INSN, que es un centro especializado en enfermedades inmunológicas que recibe a pacientes de todo el país.
Se revisaron los registros electrónicos de las citometrías de flujo constituyendo dicho registro nuestro marco muestral y llegando a 426 pacientes registrados por resultado de citometría de flujo realizada. Se seleccionaron consecutivamente a los resultados de los pacientes a los cuales se les indicó el recuento poblacional linfocitario con un diagnóstico presuntivo de IDP: agammaglobulinemias, inmunodeficiencia común variable, inmunodeficiencias combinadas, llegando a un total de 316 casos. Posteriormente se revisaron las historias clínicas de todos pacientes con sospecha de IDP y se seleccionó en base a los criterios de inclusión y exclusión, teniendo así un total de 261 casos.
Los criterios de inclusión fueron: pacientes menores de 18 años con sospecha clínica de inmunodeficiencias primarias utilizando los criterios de la Jeffrey Modell Foundation (JMF) (10); es decir, la presencia de dos o más infecciones bacterianas sistémicas o graves (por ejemplo, sepsis, osteomielitis, meningitis); tres o más infecciones bacterianas graves de los tejidos blandos respiratorias o documentadas (por ejemplo, celulitis, otitis media de drenaje, linfadenitis) dentro de un año; infecciones que ocurren en sitios inusuales (por ejemplo, abscesos hepáticos o cerebrales); infecciones con patógenos comunes de la infancia pero con gravedad inusual; infecciones oportunistas (neumonía por Pneumocystis jirovecii); aftas persistentes en pacientes mayores de un año de edad; antecedentes familiares de IDP.
De esta forma 316 casos fueron pacientes pediátricos con sospecha clínica de IDP con indicación de recuento poblacional linfocitario. Se excluyó a 55 casos por tratarse de inmunodeficiencias secundarias (neoplasias malignas, terapia reciente con esteroides o inmunosupresores).
Se consideró en la revisión de las historias clínicas a los pacientes con evaluaciones por médicos inmunólogos clínicos y presunción diagnóstica refrendada por los mismos. La determinación de las subpoblaciones linfocitarias se estableció a partir de la revisión de los registros de resultados de las citometrías de flujo realizadas a pacientes con sospecha de IDP en el Laboratorio de Inmunoalergia del CERNAAI (INSN) mediante el citómetro Fas-CANTO II. Todos los datos se recolectaron en una ficha de recolección previamente diseñada que consideró las siguientes variables: edad, género, especialidad de procedencia, valores aumentados, disminuidos o normales de TCD3+, TCD4+, TCD8+, BCD19+, NK16/56+ y relación CD4+/ CD8+, según las categorías de Comans-Bitter et al., y sus valores de referencia (Ver material suplementario) (11).
Se realizó un análisis descriptivo utilizando SPSS Statistics for Windows, versión 20.0 (SPSS Inc., Chicago, Ill., USA), expresando datos cualitativos con frecuencias absolutas y porcentajes, y datos cuantitativos con media y desviación estándar. Todos los recuentos absolutos de linfocitos se clasificaron como recuento normal o bajo para la edad, de acuerdo con los valores normales propuestos para Comans-Bitter et al. (11).
Se mantuvo la confidencialidad de toda información personal al recolectarse la información de manera anónima. El registro de información fue llevado a cabo por uno de los autores especialista en inmunología clínica. El estudio fue aprobado por el Comité de Ética en Investigación del Instituto Nacional de Salud del Niño, Breña (CL13-14).
HALLAZGOS
Se describen los resultados de 261 historias clínicas correspondientes a pacientes menores de 18 años con sospecha de IDP. El 54,8% fueron varones y la edad media de los pacientes fue de 4,37 ± 4,3 años. La mayoría procedían del servicio de Inmunología (118/261) y los pacientes de otros servicios fueron evaluados mediante interconsulta por médicos inmunólogos clínicos (Tabla 1). El recuento poblacional linfocitario para los 261 pacientes obtuvo linfopenia con linfocitos T CD3+ disminuidos en el 13,4% (35/261). Por su parte, hubo disminución de linfocitos T CD3+CD4+ en el 17,6% (46/261) de pacientes y T CD3+CD8+ en el 9,9% (26/261). Los linfocitos T NK CD16/56+ se hallaron disminuidos en 25/261 (9,6%) de pacientes. La relación CD4+/ CD8+ estuvo disminuída en 76/261 (29,1%) de pacientes (Tabla 2).

El recuento de linfocitos B se halló por debajo de valores normales en 51 pacientes; sin embargo, se halló disminuido en 34/261 (13%) de los pacientes y ausente en 17/261 (6,5%) de los pacientes. El compromiso de linfocitos B CD19+ fue la alteración más frecuentemente documentada.
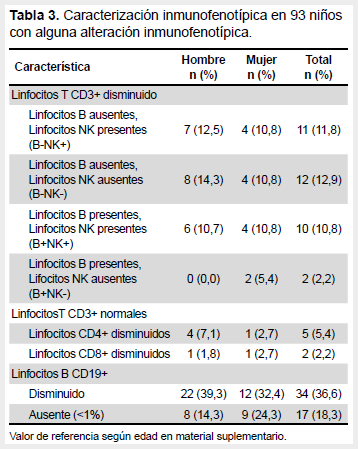
Se categorizaron a los pacientes en base a inmunofenotipos compatibles con inmunodeficiencias primarias del total de pacientes, 93/261 (35,6%) tuvieron alguna alteración inmunofenotípica (Tabla 3). Las inmunodeficiencias combinadas severas se corresponden con linfopenia; es decir, linfocitos T CD3+ disminuidos, en ese contexto tenemos cuatro inmunofenotipos: TCD3+B-NK+ que corresponden a 11/93 (11,8%) de los pacientes con alguna alteración inmunofenotípica; TCD3+B-NK-con 12/93 (12,9%); TCD3+B+NK+ con 10/93 (10,7%) y TCD3+B+NK- con 2/93 (2,2%) de los casos con alguna alteración inmunofenotípica. Las inmunodeficiencias combinadas se corresponden con TCD3+ normales pero que a la vez pueden cursar con linfocitos T CD4+ disminuidos como se observó en 5/93 (5,4%) de los casos y T CD8+ disminuido como se observó en 2/93 (2,2%) de pacientes. La inmunodeficiencia común variable inmunofenotípicamente se representa por linfocitos B CD19+ disminuidos; es decir, por debajo del valor normal para la edad pero por encima del 1%. Ello fue evidente en 34/93 (36,5%) de los casos. En el contexto de las agammaglobulinemias, se representan inmunofenotípicamente con un recuento de linfocitos B CD19+ ausentes; es decir cuando estos se hallan por debajo del 1%. Ello se observó en 17/93 (18,3%) de los casos.

DISCUSIÓN
Con respecto a los resultados de la citometría de flujo, las alteraciones más frecuentes se hallaron en el fenotipo con disminución o ausencia de linfocitos B CD19+. La deficiencia aislada de linfocitos B más importante se halla en el contexto de las agammaglobulinemias primarias donde los recuentos están ausentes, y en ICV tipo 1 donde el recuento de linfocitos B CD19+ se halla disminuído. La ICV fue el inmunofenotipo con mayor frecuencia en esta serie, hallazgo similar a lo reportado por Modell V, et al. (10).También se encuentra deficiencia de linfocitos B en las inmunodeficiencias combinadas, como la deficiencia de adenosin deasminasa y la deficiencia de RAG1 y RAG2, pero se acompañan de deficiencias en linfocitos T y/o NK (11).
Entre los pacientes con alguna alteración fenotípica, más de la tercera parte tuvieron una disminución en linfocitos T CD3+, porcentaje dentro del rango descrito en una serie en Irán (12). El hallazgo de linfocitos T CD3+ disminuidos, es importante debido a que cualquier defecto que altere el desarrollo de linfocitos T, provocaría una disminución en la inmunidad celular por constituir, de entre el 70% al 90% de los linfocitos circulantes (13). En ese sentido, la mayoría de pacientes con IDP por alteración de linfocitos T CD3+ tienen presentaciones clínicas severas en los primeros días de vida, con infecciones raras y oportunistas.
La disminución de linfocitos T CD3 (+) CD4 (+) fue poco frecuente en nuestra serie. Esta característica esta presente en inmunofenotipos de diversas IDP como la deficiencia MHC II, inmunodeficiencia combinada severa ligada al cromosoma X (SCID-X) con paso transplacentario de linfocitos T de origen materno, enfermedad candidiásica mucocutánea crónica, ataxia-telangiectasia, entre otras (14).
En este estudio se determinó que la tercera parte de pacientes con sospecha de IDP presentó disminución de la relación CD4/ CD8, hallazgo asociado con anemia ferropénica según una serie (15 ).Asimismo, no se consideró el estudio de anemia en estos pacientes; sin embargo, sería importante abordar tal aspecto considerando que el hierro es esencial para el desarrollo funcional normal del sistema inmunológico, por ello su deficiencia puede resultar en una respuesta inmune inadecuada (16). Estudios en pacientes pediátricos infectados con el virus de la inmunodificiencia humana, inmunodeprimidos desde el nacimiento, describen que la disminución de la relación CD4/CD8 se asocia como marcador de senescencia inmunológica y de riesgo de infección virales crónicas, al ser un marcador de inmunoactivación (17). Los niveles altos de la relación de CD4/CD8 también se han asociado con el desarrollo de la autoinmunidad en pacientes con ICV (18).
El laboratorio del centro donde se realizó el estudio no cuenta con pruebas de diagnóstico molecular ni genético, por lo que para precisar el diagnóstico específico, las muestras son enviadas a laboratorios del exterior, ello podría contribuir al retraso diagnóstico que para Veramendi-Espinoza et al. (19), se estima en una mediana de 12 meses, con un rango intercuartil muy amplio de 5,1 a 30,3 meses. Por otra parte, Córdova (20), en la seguridad social del Perú, reportó 15 pacientes con diagnóstico de IDP con una mediana de retraso en el diagnóstico de seis años y siete meses (rango intercuartil: 1-36 años). Este retraso en el diagnóstico podría implicar costos mayores al sistema de salud peruano al no iniciarse un tratamiento oportuno.
Una limitación importante de nuestro estudio es la poca disponibilidad de datos clínicos, ello debido a que la recolección de datos se realizó de fuentes secundarias. Por otra parte, a pesar de haberse realizado el estudio en un centro de referencia, es difícil establecer si los pacientes incluidos son representativos de todos los pacientes peruanos con sospecha de IDP.
De este modo, hemos podido aproximarnos de modo preliminar al diagnóstico de IDP en base a inmunofenotipos compatibles en casi la tercera parte de pacientes con sospecha clínica. En ese contexto, se concluye que el inmunofenotipo de inmunodeficiencia común variable fue más frecuente (36,6%), seguido de agammaglobulinemias (18,3%). Ambos inmunofenotipos son defectos en la producción de anticuerpos.
Contribuciones de autoría: EMB y RIM concibieron y plantearon el proyecto de investigación. DGG y DMQ diseñaron el proyecto y realizaron el análisis estadístico. DGG, DMQ y JCH participaron en la elaboración del artículo. MLT, WCC, RML y CGC participaron activamente en la revisión del proyecto y del artículo final. Todos los autores contribuyeron activamente desde la concepción del problema hasta la redacción del artículo.
Fuentes de financiamiento: autofinanciado. Conflictos de interés: los autores declaran no tener conflictos de interés.
Material suplementario: Disponible en la versión electrónica de la RPMESP.
REFERENCIAS BIBLIOGRÁFICAS
1. Ballow M. Primary immunodeficiency disorders: Antibody deficiency. J Allergy Clin Immunol. 2002;109(4):581-91. doi: 10.1067/mai.2002.122466.
2. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova J, et al. Primary Immunodeficiency Diseases Worldwide: More Common than Generally Thought. J Clin Immunol. 2013;33(1):1-7. doi: 10.1007/s10875-012-9751-7.
3. Mendoza-Quispe D, García-Gomero D, Córdova-Calderón W. Diagnóstico situacional de las inmunodeficiencias primarias:aproximación preliminar. Rev Peru Med Exp Salud Publica. 2017;34:346-7. doi: 10.17843/rpmesp.2017.342.2687.
4. Costa-Carvalho B, González-Serrano M, Espinosa-Padilla S, Segundo G. Latin American challenges with the diagnosis and treatment of primary immunodeficiency diseases. Expert Rev Clin Immunol. 2017;13(5):483-9. doi: 10.1080/1744666X.2017.1255143.
5. Richardson AM, Moyer AM, Hasadsri L, Abraham RS. Diagnostic Tools for Inborn Errors of Human Immunity (Primary Immunodeficiencies and Immune Dysregulatory Diseases). Curr Allergy Asthma Rep. 2018;18(3):19. doi: 10.1007/s11882-018-0770-1.
6. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2018;38(1):129-43. doi: 10.1007/s10875-017-0465-8.
7. Illoh OC. Current Applications of Flow Cytometry in the Diagnosis of Primary Immunodeficiency Diseases. Arch Pathol Lab Med. 2004;128(1):23-31. doi: 10.1043/1543-2165 (2004)128<23:CAOFCI>2.0.CO;2.
8. Azizi G, Ghanavatinejad A, Abolhassani H, Yazdani R, Rezaei N, Mirshafiey A, et al. Autoimmunity in primary T-cell immunodeficiencies. Expert Rev Clin Immunol. 2016;12(9):989-1006. doi: 10.1080/1744666X.2016.1177458.
9. Gallo V, Dotta L, Giardino G, Cirillo E, Lougaris V, D’Assante R, et al. Diagnostics of Primary Immunodeficiencies through Next-Generation Sequencing. Front Immunol. 2016;7:466. doi: 10.3389/fimmu.2016.00466.
10. Modell V, Gee B, Lewis D, Orange J, Roifman C, Routes J, et al. Global study of primary immunodeficiency diseases (PI)—diagnosis, treatment, and economic impact: an updated report from the Jeffrey Modell Foundation. Immunol Res. 2011;51(1):61-70. doi: 10.1007/s12026-011-8241-y.
11. Comans-Bitter WM, de Groot R, van den Beemd R, Neijens HJ, Hop WC, Groeneveld K, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997;130(3):388-93.
12. Mohammadinejad P, Mirminachi B, Sadeghi B, Movahedi M, Gharagozlou M, Mohammadi J, et al. Distribution of primary immunodeficiency disorders diagnosed in a tertiary referral center, Tehran, Iran (2006-2013). Iran J Immunol IJI. 2014;11(4):282-91. doi: IJIv11i4A7.
13. Edgar JD. T cell immunodeficiency. J Clin Pathol. 2008;61(9):988-93. doi: 10.1136/jcp.2007.051144.
14. Marcus N, Stauber T, Lev A, Simon AJ, Stein J, Broides A, et al. MHC II deficient infant identified by newborn screening program for SCID. Immunol Res. 2018;66(4):537-42. doi: 10.1007/s12026-018-9019-2.
15. Aly S, Fayed H, Ismail A, Hakeem G. Assessment of peripheral blood lymphocyte subsets in children with iron deficiency anemia. BMC Pediatr. 2018;18(1):49. doi: 10.1186/s12887-018-0990-5.
16. Weiss G. Iron, infection and anemia--a classical triad. Wien Klin Wochenschr. 2002;114(10-11):357-67.
17. Alvarez P, Mwamzuka M, Marshed F, Kravietz A, Ilmet T, Ahmed A, et al. Immune activation despite preserved CD4 T cells in perinatally HIV infected children and adolescents. PLoS One. 2017;12(12):e0190332. doi: 10.1371/journal.pone.0190332.
18. Abolhassani H, Amirkashani D, Parvaneh N, Mohammadinejad P, Gharib B, Shahinpour S, et al. Autoimmune phenotype in patients with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2013;23(5):323-9.
19. Veramendi-Espinoza L, Zafra-Tanaka JH, Pérez-Casquino GA, Córdova-Calderón WO. Diagnostic Delay of Primary Immunodeficiencies at a Tertiary Care Hospital in Peru- Brief Report. J Clin Immunol. 2017;37(4):383-7. doi: 10.1007/s10875-017-0398-2.
20. Córdova W. Características clínicas y epidemiológicas de las inmunodeficiencias primarias en el Hospital Guillermo Almenara Irigoyen entre los años 2000 y 2010. [Tesis de postgrado]. Lima, Perú: Universidad Nacional Mayor de San Marcos; 2012.
Correspondencia: David Santiago García Gomero.
Dirección: Calle Quipán 157 Urb. Tahuantinsuyo-Independencia. Lima, Perú.
Teléfono: +51926641560
Correo electrónico: dsantg.g@gmail.com
Recibido: 07/11/2018
Aprobado: 21/08/2019
En línea: 23/09/2019