
ORIGINAL BREVE
Detección de mutaciones causantes de distrofia muscular de Duchenne/Becker: reacción en cadena de la polimerasa multiplex vs. amplificación múltiple dependiente de ligación por sondas
Detection of mutations causing Duchenne and Becker muscular dystrophies: Multiplex polymerase chain Reaction vs. Multiplex Ligation dependent Probe Amplification
Francia DP. Huamán-Dianderas1,a, María Luisa Guevara-Fujita1,b, Diana Rojas Málaga2,c, Alejandro Estrada-Cuzcano3,d, Ricardo Fujita1,e
1 Centro de Investigación de Genética y Biología Molecular, Instituto de Investigación, Facultad de Medicina Humana, Universidad de San Martín de Porres. Lima, Perú.
2 Laboratório de Genética y Biologia Molecular, Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Brasil.
3 Medical Genetics Laboratory, INSERM U1112, Institute of Medical Genetics of Alsace, University of Strasbourg, Strasbourg Medical School. Strasbourg, France.
a Bióloga, magister en Investigación Clínica; b bióloga, doctora en Ciencias con mención en Bioquímica y Biología Molecular; c bióloga, doctora en Genética y Biología Molecular; d biólogo, PhD en Medical Sciences; e biólogo, doctor en Ciencias con mención en Bioquímica y Biología Molecular.
RESUMEN
Las distrofias musculares de Duchenne/Becker son enfermedades raras que reciben poca atención en nuestro medio. El objetivo del presente estudio fue implementar la técnica de amplificación múltiple dependiente de ligación por sondas (MLPA) y demostrar que tiene ventajas sobre la técnica de reacción en cadena de la polimerasa multiplex (PCR-multiplex). Se analizaron muestras de 40 individuos con diagnóstico presuntivo de distrofia muscular de Duchenne/Becker, primero por PCR-multiplex y luego por MLPA. Con la PCR-multiplex se detectaron 15 individuos con deleciones causales y con la técnica MLPA se logró diagnosticar a 21 individuos, cuatro duplicaciones y 17 deleciones. En conclusión, la técnica MLPA logra detectar mutaciones de tipo deleción y duplicación de exones, consiguiendo un mayor número de diagnósticos moleculares por alteraciones en el gen DMD.
Palabras claves: Distrofina; Reacción en cadena de la polimerasa multiplex; Amplificación múltiple dependiente de ligación por sondas; Diagnóstico genético (fuente: DeCS BIREME).
ABSTRACT
Duchenne and Becker muscular dystrophies are rare diseases that receive limited attention in our field. The objective of this study was to implement the Multiplex Ligation-dependent Probe Amplification technique (MLPA) and to demonstrate that it has advantages over the Multiplex Polymerase Chain Reaction (Multiplex PCR) technique. Samples from 40 individuals with a presumptive diagnosis of Duchenne and Becker muscular dystrophies were analyzed: first by Multiplex PCR and then by MLPA. Fifteen individuals with causal deletions were detected with Multiplex PCR, while the MLPA technique was able to diagnose 21 individuals, four duplications, and 17 deletions. In conclusion, the MLPA technique can detect mutations of the exon deletion and duplication type, yielding a larger number of molecular diagnoses due to alterations in the DMD gene.
Keywords: Dystrophin; Multiplex polymerase chain reaction; Multiplex ligation-dependent probe amplification; Genetic diagnosis (source: MeSH NLM).
INTRODUCCIÓN
Todos los tipos de distrofia muscular son enfermedades que producen debilidad muscular, las más comunes son la distrofia muscular de Duchenne (DMD) y la de Becker (BMD) y son causadas por variaciones del gen DMD, uno de los más grandes del genoma humano (2.4Mb), compuesto por 79 exones y siete promotores, ubicado en la región Xp21.1 (1).Dicho gen se expresa en músculo esquelético, cardiaco, células neuronales y músculo liso, y se traduce en distrofina, proteína que forma parte del complejo proteico asociado a distrofina (DAP), cuya función es estructural, pues debe estabilizar el sarcolema y proteger a las fibras musculares del daño y necrosis ocasionados por la contracción a largo plazo (1,2).
La DMD (OMIM 310200) es la más frecuente y tiene una incidencia de 1 por cada 3500 niños nacidos vivos (3). Tiene un pronóstico grave y expectativa de vida reducida. La BMD (OMIM 300376) tiene una incidencia de 1 por cada 30 000 niños nacidos vivos y tiene una forma más leve de presentación, con pronóstico de vida cercano a los 60 años (3).
Los casos de DMD son más graves debido a que la distrofina se presenta muy reducida, no funcional o en ausencia total, debido teóricamente a variaciones fuera del marco de lectura en el gen DMD (3,4). Mientras que en los casos de DMB, las variaciones se encuentran dentro del marco de lectura, expresando una proteína incompleta pero funcional y por lo tanto con mejor pronóstico para el paciente (3,4).
En Perú, como en la mayoría de países en vías de desarrollo, el diagnóstico para DMD/DMB consta básicamente de la identificación de diferentes signos clínicos, como dificultad para caminar, hipotonía, atrofia muscular, signo de Gowers, escoliosis, contracturas articulares, y en cuadros avanzados, insuficiencia respiratoria y/o cardiaca; este diagnóstico se apoya en el nivel de creatina quinasa (CPK), elevada en varones si es mayor a 174 u/L; y en la electromiografía (EMG), que mide la actividad eléctrica de los músculos y distingue entre patrón neuromuscular normal, miopático o neuropático (4–7).
Como DMD/DMB son enfermedades monogénicas, todas las técnicas de diagnóstico confirmatorias están relacionadas a la detección directa de la distrofina in situ, como el análisis inmunohistoquímico o Western blot (4-7), y a la identificación de las variantes patológicas del mismo gen DMD, usando la técnica de reacción en cadena de la polimerasa multiplex (PCRmultiplex) (Test de Chamberlain, Beggs o Kunkel) que detecta sólo presencia/ausencia de unos 9 a 11 exones por reacción (5), o la técnica de amplificación múltiple dependiente de ligación por sondas (MLPA) que detecta la presencia/ausencia y dosis de los 79 exones y el promotor Dp427c en dos reacciones (8). Además, la secuenciación Sanger detecta pequeñas mutaciones en todas las regiones del gen DMD (una reacción por región) y la secuenciación de nueva generación (NGS) detecta la ausencia de grandes regiones (multiexónicas) y pequeñas mutaciones en todo el gen en una sola reacción (9,10).
El objetivo del presente estudio fue implementar la técnica MLPA para el diagnóstico de DMD/DMB y demostrar su ventaja sobre la PCR-multiplex, comúnmente utilizada en algunos centros hospitalarios del Perú.
EL ESTUDIO
El presente estudio es de tipo descriptivo prospectivo, comprende la primera parte de un proyecto de investigación denominado «Diagnóstico genético molecular en pacientes con Distrofia Muscular de Duchenne (DMD) y Becker (BMD) en Perú», que busca caracterizar genéticamente a 300 pacientes por diferentes técnicas moleculares durante tres años. El proyecto está financiado por la Facultad de Medicina Humana de la Universidad de San Martín de Porres.
Durante el primer año se reclutaron 60 pacientes provenientes del Servicio de Genética y Errores Innatos del Metabolismo del Hospital del Niño y del Servicio de Genética del Departamento de Especialidades Médicas del Hospital Edgardo Rebagliati Martins. Para este estudio se consideraron a 40 pacientes y a dos familias nucleares (de los únicos dos pacientes que aceptaron toma de muestra). Además, se utilizaron diez controles masculinos sanos del banco de ADN del Centro de Investigación de Genética y Biología Molecular (CIGBM), tal como se recomienda en el protocolo de MLPA-DNA versión MDP-v003 de MRC-Holland (11).
Se incluyeron a pacientes peruanos, varones (por ser los afectados en una enfermedad ligada al X), menores de 18 años, diagnosticados clínicamente con distrofia muscular que presentan pruebas de apoyo al diagnóstico, CPK elevada y electromiografía que evidencia un patrón miopático. Se excluyeron pacientes con información clínica incompleta.
Se extrajo ADN (aproximadamente 500 uL) a partir de 3 mL de sangre periférica, siguiendo el protocolo descrito por Miller et al. («salting out») (12) estandarizado en el CIGBM.
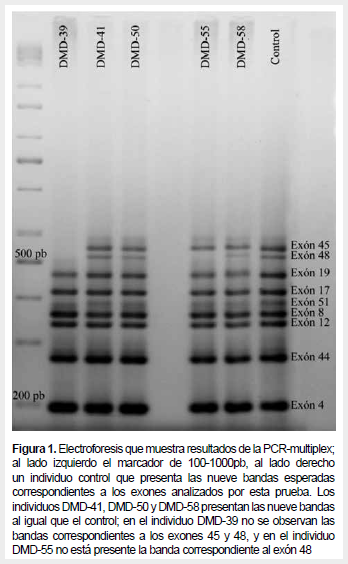
Para la PCR-multiplex de nueve exones: exón 4 (186pb), exón 44 (268pb), exón 12 (331pb), exón 8 (360pb), exón 51 (328pb), exón 17 (417pb), exón 19 (459pb), exón 48 (505pb) y exón 45 (547pb) la amplificación se realizó según el protocolo descrito por Rojas, et al. (13). La detección mediante electroforesis utilizó agarosa de alta sensibilidad al 4%, con un programa de 30 voltios por 20 min, 60 voltios por 40 min, 90 voltios por una hora y 120 voltios por dos horas. La tinción fue con bromuro de etidio y las fotos se consiguieron con un fotodocumentador Biorad Gel Doc XR.
La MLPA consta de cuatro pasos: denaturación del ADN, hibridación con las sondas, ligación de estas y amplificación de los fragmentos que previamente han hibridado y ligado. Se realizó según el protocolo de MLPA-DNAversión MDP-v003 (11), con pequeñas modificaciones (la hibridación duró de 16 a 20 horas y la ligación de las sondas duró 40 minutos). Se utilizaron los mixes de sondas comerciales: P034 y P035 (MRC-Holland), que en conjunto analizan los 79 exones del gen DMD y el promotor Dp427c. Los amplificados se corrieron por electroforesis capilar en un equipo ABI-3500, 1 uL de muestra más 9 uL de formamida HiDi y 0,2 uL de Liz 500. Se usó un capilar de 50 cm y POP 7, y las condiciones fueron de 1,6 KV de voltaje de inyección y 15 s de tiempo de inyección.
Siguiendo lo recomendado en el inserto de los mixes de sondas P034 y P035 se procedieron a confirmar las deleciones de un solo exón, mediante una reacción en cadena de polimerasa (PCR) de dos exones a la vez (el exón que tiene supuestamente la deleción y un exón control adicional).
Los datos extraídos de la electroforesis capilar se analizaron mediante el software libre Coffalyser.Net v.140721.1958 (www.mlpa.com), también recomendado por la MRC-Holland (11). Este análisis se realizó sobre el radio del DQ (dosaje de la fluorescencia captada por cada fragmento). Se evaluó la presencia o ausencia de cada fragmento y la dosis en que se presentan, en comparación con las muestras control.
Antes de la obtención de la muestra, los padres o apoderados de los participantes firmaron voluntariamente un consentimiento informado. El protocolo de investigación, así como el formato de consentimiento fue aprobado por el Comité Institucionalde Ética en Investigación de la Universidad San Martín de Porres, el cual respeta las normas éticas concordantes con la declaración de Helsinki (actualizada al 2013).
HALLAZGOS
De las 40 muestras analizadas mediante PCR-multiplex, 15 (37,5%) presentaron deleciones de uno o más exones del gen DMD, causales de distrofia muscular (Tabla 1). Estas deleciones se evidencian por la falta de las bandas correspondientes a determinados exones en comparación con el control (Figura 1). En las pruebas realizadas a las madres y/o hermanas de dos pacientes, no se encontró ninguna variación del patrón de bandas con respecto al control.
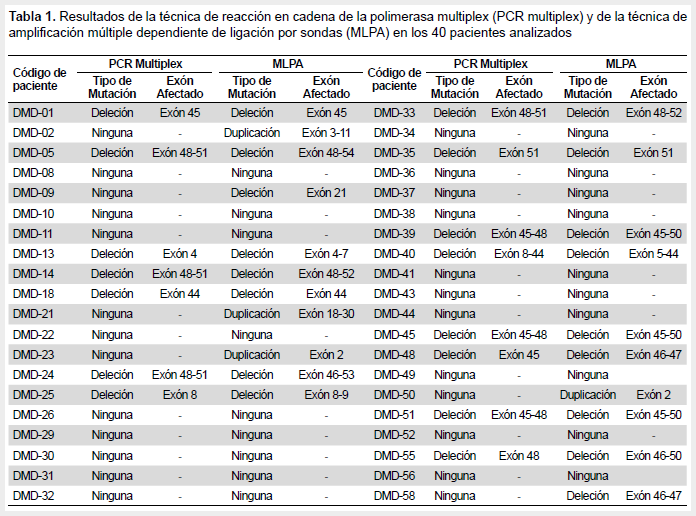
De las 40 muestras analizadas mediante MLPA, 21 (52,5%) fueron diagnosticados, de los cuales 17 (42,5%) corresponde a deleciones y 4 (10%) a duplicaciones de uno o más exones del gen DMD (Tabla 1). Además, las cuatro deleciones de un solo exón fueron verificadas por una PCR de dos exones. Al realizarse esta prueba a los familiares (madres y/o hermanas) de los mismos individuos escogidos anteriormente, estas resultaron ser portadoras.
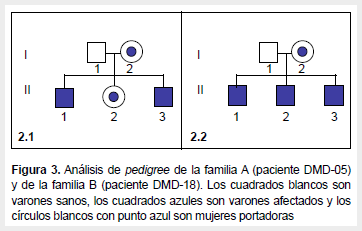
En el pedigree de la familia A, mediante PCR-multiplex el paciente DMD-05 (II-1) tiene deleciones de los exones 48 y 51 del gen DMD (Figura 2), y mediante MLPA se determinó que la deleción realmente abarca desde el exón 48 hasta el 54. El paciente tiene un hermano menor (II-3) que también presenta la mutación confirmada por ambas técnicas, mientras que la hermana (II-2) y la madre (I-2) son portadoras de la mutación, detectadas sólo por MLPA (Figura 3).
En el pedigree de la familia B, mediante PCR-multiplex el paciente DMD-18 (II-1) tiene una deleción del exón 44 del gen DMD y mediante la técnica MLPA se confirmó que sólo presenta la deleción de ese exón. Tiene dos hermanos menores (II-2 y II-3) que también presentan la misma mutación confirmada por ambas técnicas, mientras que la madre (I-2) es portadora de dicha mutación, detectada sólo por MLPA (Figura 3).
DISCUSIÓN
Actualmente en los centros hospitalarios del Perú, son poco recomendables las pruebas inmunohistoquímicas o de Western blot, ya que para ellas son necesarias muestras de biopsia muscular, consideradas muy invasivas. La PCR-multiplex se encuentra implementada desde hace varios años, la MLPA y el NGS no son de uso rutinario para el diagnóstico y la secuenciación Sanger se utiliza para el descarte de una mutación ya conocida, ya que implica un mayor gasto de tiempo y dinero (6,9,20).
En el presente estudio se demuestra que la MLPA diagnostica 15% más de casos que la PCR-multiplex; además, la MLPA confirma el 100% los casos detectados por PCR-multiplex y de estos un 80% se amplía y consigue un diagnóstico más exacto en cuanto al verdadero tamaño de la mutación. Estas ventajas permiten un mejor diagnóstico de la distrofinopatía que padece el paciente, y permite brindar un correcto pronóstico de la enfermedad. Estos resultados se corresponden con lo reportado por Schwartz & Duno (14), Lalic, et al. (15), Janssen, et al. (16), Lai, et al. (17) y Guo, et al. (18), quienes también confirmaron los resultados. Esto permite inferir que la técnica MLPA, para el análisis del gen DMD, es un método reproducible, fácil de manejar y más sensible que la PCR-multiplex.
Para citar un ejemplo, en el paciente DMD-24, la PCR-multiplex detecta la ausencia de los exones 48 y 51. Con MPLA se descubre que la verdadera mutación comprende una deleción desde el exón 46 hasta el exón 53. Esto es muy importante, ya que de darse el resultado de PCR-multiplex como una deleción del exón 48 al 51 no se altera el marco de lectura y por lo tanto se infiere que la proteína a formarse estaría alterada pero funcional, tratándose de un posible caso de DMB. Sin embargo, al conocerse que la verdadera mutación es desde el exón 46 hasta el 53 sí se altera el marco de lectura de la proteína parando tempranamente la traducción en el exón 45 por lo que se infiere una proteína muy pequeña que no presentaría su extremo terminal C. Esto evitaría la interacción de la distrofina con los miembros del DAP, ocasionando el cuadro clínico de Duchenne.

Otro punto importante es que a diferencia de la PCR-multiplex, que sólo es capaz de detectar deleciones, con la MLPA se diagnostican tanto deleciones como duplicaciones, que en el presente estudio se encuentran en un 43% y 10%, respectivamente. Estos porcentajes son diferentes a los reportados por diferentes autores en distintas poblaciones, aunque la más resaltante es la base de datos internacional LOVD (Leiden Open Variation Database) que, actualizada a mayo 2019, con datos de 14 260 variantes patogénicas en 37 626 casos indica que las mutaciones patológicas en el gen DMD se presentan como deleciones en un 64%, duplicaciones en un 12% y pequeñas mutaciones (de uno o más nucleótidos) en el 23% de los casos (19).
Adicionalmente, la MLPA, al cuantificar el número de copias, también puede detectar mujeres portadoras de la enfermedad (20). En familias con un individuo afectado, puede estudiarse también a las mujeres (hijas o tías maternas) para saber si son portadoras de la mutación que causa la enfermedad. Esto reviste importancia porque se puede brindar asesoría genética a la familia, a fin de que ésta tome decisiones informadas respecto a futuros embarazos. Esto debido a que una mujer portadora, tiene un 50% de probabilidad de heredar la mutación; por lo tanto, si tiene un hijo varón este tiene un 50% de probabilidad de estar afectado y si es mujer, un 50% de ser portadora (10).
En conclusión, la técnica de MLPA diagnostica con mayor precisión que la PCR-multiplex los casos de DMD/DMB, detecta tanto deleciones como duplicaciones de cualquiera de los 79 exones del gen DMD, e identifica mujeres portadoras asintomáticas. Aunque el tamaño de la muestra sea una limitante, estos resultados propician la implementación de la técnica MLPA en el CIGBM, y podemos recomendarla para diagnóstico molecular de DMD/DMB. Esta prueba es de fácil reproducción en un laboratorio que cuente con un termociclador y un secuenciador, y aunque es más costosa que la PCRmultiplex, tiene mayor sensibilidad y permite la detección exacta del tamaño de las deleciones/duplicaciones en este gen.
Contribuciones de los autores: MLGF elaboró el proyecto principal; FDPHD y MLGF realizaron el diseño experimental, trabajo experimental, el análisis e interpretación de datos; MLGF, DRM y AEC contribuyeron con el reclutamiento de pacientes y la obtención de muestras; FDPHD realizó el primer borrador del artículo. Todos los autores contribuyeron en la revisión y edición de la versión a publicar.
Fuentes de financiamiento: El presente artículo forma parte del proyecto de investigación, «Diagnóstico genético molecular en pacientes con Distrofia Muscular de Duchenne (DMD) y Becker (BMD) en Perú», financiado por la Facultad de Medicina Humana de la Universidad de San Martín de Porres.
Conflictos de interés: Los autores declaran no presentar ninguna relación, condición o circunstancia de índole económico o institucional, que pueda afectar la objetividad de la interpretación del artículo.
REFERENCIAS BIBLIOGRÁFICAS
1. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–40. Doi: 10.1016/S1474-4422(03)00585-4.
2. Blake DJ, Weir A, Newey SE, Davies KE. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol Rev. 2002;82(2):291-329. Doi: 10.1152/physrev.00028.2001.
3. Silva CT, Fonseca DJ, Mateus H, Contreras N, Restrepo CM. Distrofia muscular de Duchenne y Becker, una visión molecular. Acta Médica Colomb. 2005;30:113.
4. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. Doi: 10.1016/S14744422(09)70271-6.
5. Bushby KMD, Anderson LVB, eds. Muscular Dystrophy: Methods and Protocols. Totowa, NJ: Humana Press; 2001.
6. Falzarano M, Scotton C, Passarelli C, Ferlini A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules. 2015;20(10):18168-84. Doi: 10.3390/molecules201018168.
7. Montejo-Pujadas Y, Zaldívar-Vaillant T, Acevedo-López AM. Técnicas diagnósticas descritas en el estudio de la distrofia muscular de Duchenne/Becker. Rev Neurol. 2002;34:278-81. Doi: 10.33588/rn.3403.2001286.
8. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57–e57. Doi: 10.1093/nar/gnf056.
9. Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975,94(3):441-8. Doi: 10.1016/0022-2836(75)90213-2.
10. Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145-51. Doi: 10.1136/jmedgenet-2015-103387.
11. MRC-Holland. MLPA DNA protocol version MDP-v003 [Internet]. Amsterdam: MRC-Holland; 2013. Disponible en: www.mlpa.com.
12. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988;16(3):1215.
13. Rojas D, Narvaja ME, Rivas L, Guevara-Fujita ML, Castañeda C, Fujita R. Implementación de la Prueba del Multiplex PCR para el Gen DMD en Pacientes con sospecha de Distrofia Muscular de Duchenne/Becker y la identificación de una deleción de los exones 48-5. Rev Horiz Med. 2012;12(3):8-15. Doi: 10.24265/horizmed.
14. Schwartz M, Dunø M. Multiplex ligation-dependent probe amplification is superior for detecting deletions/duplications in Duchenne muscular dystrophy: Multiplex ligation-dependent probe amplification. Clin Genet. 2004;67(2):189-91. Doi: 10.1111/j.13990004.2004.00382.x.
15. Lalic T, Vossen RH, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13(11):1231–4. Doi: 10.1038/sj.ejhg.5201465.
16. Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics. 2005;6(1):29-35. Doi: 10.1007/s10048-004-0204-1.
17. Lai KK, Lo IF, Tong TM, Cheng LY, Lam ST. Detecting exon deletions and duplications of the DMD gene using Multiplex Ligationdependent Probe Amplification (MLPA). Clin Biochem. 2006;39(4):367-72. Doi: 10.1016/j.clinbiochem.2005.11.019.
18. Guo R, Zhu G, Zhu H, Ma R, Peng Y, Liang D, et al. DMD mutation spectrum analysis in 613 Chinese patients with dystrophinopathy. J Hum Genet. 2015;60(8):435-42. Doi:10.1038/jhg.2015.43.
19. Aartsma-Rus A, Van Deutekom JCT, Fokkema IF, Van Ommen G-JB, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34(2):135-44. Doi: 10.1002/mus.20586.
20. Gatta V, Scarciolla O, Gaspari AR, Palka C, De Angelis MV, Di Muzio A, et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet. 2005;117(1):92-8. Doi:10.1007/s00439-005-1270-7.
Correspondencia: Francia del Pilar Huaman-Dianderas
Dirección: Av. Del Pacífico 180, Parques de la Huaca 6ta etapa, Torre 4, Dpto 1001. Lima, Perú.
Teléfono: (511) 980654809
Correo electrónico: franciahd19@gmail.com
Recibido: 26/11/2018
Aprobado: 17/07/2019
En línea: 16/08/2019