
ORIGINAL BREVE
Diagnóstico genético de pacientes con agammaglobulinemia primaria atendidos en centros peruanos de tercer nivel
Genetic diagnosis of patients with primary agammaglobulinemia treated at third level peruvian centers
Edgar Matos-Benavides1,2,3,a, David García-Gomero1,2,4,b, Rosario Inocente-Malpartida1,2,4,c, Wilmer Córdova-Calderón1,2,a, Juan Aldave-Becerra2,5,a
1 Centro de Referencia Nacional de Alergia, Asma e Inmunología. Instituto Nacional de Salud del Niño. Lima, Perú.
2 Sociedad Peruana de Inmunología (SPI). Lima, Perú.
3 Universidad Nacional Mayor de San Marcos. Lima, Perú.
4 Universidad Peruana Cayetano Heredia. Lima, Perú.
5 Hospital Nacional Edgardo Rebagliati Martins. Lima, Perú.
a Médico inmunólogo clínico-alergólogo; b médico cirujano; c tecnólogo médico.
RESUMEN
Las agammaglobulinemias primarias (AP) resultan de alteraciones específicas en las células B, lo cual, conduce a baja producción de anticuerpos. La sospecha diagnóstica se establece con el antecedente de infecciones a repetición, inmunoglobulinas bajas y la ausencia linfocitos B CD19+. El diagnóstico se confirma mediante el análisis genético y la detección de una mutación ligada en el cromosoma X o autosómico recesiva o dominante. En Perú, no hay literatura sobre AP ni reportes sobre el genotipo de los pacientes con sospecha de AP. Bajo este escenario, se realizó un estudio que describió el genotipo de pacientes con sospecha de AP. Se encontraron 20 pacientes con mutaciones en el gen BTK y una mutación autosómica recesiva IGHM. Se hallaron 13 mutaciones hereditarias y siete mutaciones de novo. Se concluye que las AP son, en su mayoría, mutaciones en el gen BTK que corresponden con AP ligadas al cromosoma X.
Palabras clave: Agammaglobulinemia; Genotipo; Agammaglobulinemia Tirosina Quinasa; Perú (fuente: DeCS BIREME).
ABSTRACT
Primary agammaglobulinemia result from specific alterations in B cells, which lead to low antibody production. Diagnostic suspicion is established with a history of repeated infections, low immunoglobulins, and absence of CD19+ B lymphocytes. The diagnosis is confirmed by genetic analysis and the detection of a mutation linked to the X or autosomal recessive or dominant chromosome. In Peru, there is no literature on primary agammaglobulinemia and no reports on the genotype of patients with suspected primary agammaglobulinemia. Under this scenario, a study was performed to describe the genotype of patients with suspected primary agammaglobulinemia. Twenty (20) patients were found with mutations in the BTK gene and an autosomal recessive IGHM mutation. Thirteen (13) hereditary mutations and seven de novo mutations were found. It is concluded that the group of primary agammaglobulinemia are mostly mutations in the BTK gene, corresponding to X-linked agammaglobulinemia.
Keywords: Agammaglobulinaemia; Genotype, Bruton type agammaglobulinemia; Peru (source: MeSH NLM).
INTRODUCCIÓN
Las agammaglobulinemias primarias (AP) son un grupo de enfermedades originadas por la alteración en la producción de anticuerpos a causa de un defecto molecular ya sea intrínseco a las células B o un fracaso de las interacciones entre células B y T (1). Las AP se definen en base a un recuento de linfocitos B CD19+ menor a 1% en el contexto de un conteo bajo de anticuerpos (2).
La deficiencia de anticuerpos conduce típicamente a infecciones recurrentes, a menudo graves, de las vías respiratorias superiores e inferiores por bacterias encapsuladas, tales como Streptococcus pneumoniae y Haemophilus influenzae (3). Existen tres tipos de AP: la agammagobulinemia ligada al cromosoma X (ALX) que se manifiesta en pacientes varones y se deriva de alteraciones en el gen Bruton Tirosin Kinasa (BTK), la autosómico recesiva y la autosómica dominante los cuales se presentan en ambos sexos (4, 5).
El diagnóstico de AP se confirma con estudios genéticos que buscan identificar una mutación en el gen BTK para la ALX o al menos alguna de las seis variantes genéticas para la forma autosómica recesiva, o alguna de las dos variantes genéticas para la forma autosómico dominante (6). Se ha reportado que las ALX son las AP más frecuentes, la mayoría de estas son mutaciones hereditarias, definidas estas como aquellas que se transmiten por la línea materna, en el contexto de herencia ligada al sexo. Así lo reportó Zhang et al. quien describió el genotipo de seis pacientes con AP en China y confirmó la ALX a partir de la detección de mutaciones en exones del gen BTK, sólo reportó una mutación de novo, definida esta como aquella mutación que no es transmitida por línea materna o paterna y que ocurre espontáneamente (7). Por su parte, Esenboga et al. reportó los resultados del análisis genético de 26 pacientes con sospecha de AP y determinó mutaciones BTK en todos los pacientes, además describió una ausencia de relación entre el genotipo y el fenotipo de la enfermedad en términos de severidad (8).
En Perú, existe un infradiagnóstico de inmunodeficiencias primarias (IDP), ello debido en gran parte a las dificultades logísticas y económicas para la realización de un diagnóstico confirmatorio (9). Por su parte, los defectos en la producción de anticuerpos son las IDP más frecuentes reportadas en Perú y su tratamiento implica la utilización de inmunoglobulina humana, medicamento fundamental para alterar la historia natural de la enfermedad, evitar infecciones a repetición y hospitalizaciones (10).
El objetivo del presente estudio fue describir el genotipo de pacientes con sospecha de AP atendidos en centros del tercer nivel, este es el primer reporte peruano en el cual se llega a un diagnóstico definitivo de AP.
EL ESTUDIO
Se realizó un estudio descriptivo que incluyó a pacientes menores de 18 años con sospecha clínica de agammaglobulinemia primaria que acudieron al Centro de Referencia Nacional de Alergia e Inmunología (CERNAAI) del Instituto Nacional de Salud del Niño (INSN), pero que en muchos casos se atienden actualmente en otros centros de tercer nivel. Los pacientes pertenecen a un registro peruano de pacientes con inmunodeficiencias primarias, previamente reportado (10). Se incluyeron pacientes menores de 18 años con diagnóstico de agammaglobulinemias primarias establecido por un inmunólogo clínico en base a criterios de la International Union of Immunological Societies (IUIS) con cuadro clínico compatible y de linfocitos B CD19+ ausentes (1) entre el 1 de junio de 2011 al 1 de junio de 2014. Se excluyeron a pacientes sin clínica de agammaglobulinemia primaria, con inmunodeficiencia secundaria o que no hayan aceptado su participación a través del consentimiento informado.
Se estableció contacto con los padres o el apoderado del paciente para la realización del consentimiento informado siguiendo todos los lineamientos del tratado de Helsinsky. Hubo dos consentimientos informados, uno para la participación del paciente en el estudio y otro para la toma de muestra de la madre del paciente. Esta muestra fue tomada para definir si la madre fue portadora de la mutación.
Las muestras de sangre se extrajeron el día de la entrevista. Se obtuvo 5 ml de muestra de sangre en tubos de sistema al vacío con EDTA, previa asepsia. Se etiquetó la muestra según el número de ficha. Se envió todas las muestras para su procesamiento en un laboratorio externo por el método PCR y secuenciación directa. Las muestras de sangre para la secuenciación no requirieron ningún procedimiento especial para su conservación.
El análisis estadístico se realizó mediante el programa estadístico SPSS versión 24 (IBM Corp., Armonk, NY, EE. UU.). Para la descripción de las variables cualitativas se utilizaron las frecuencias y porcentajes. Se aplicó la prueba de normalidad de Shapiro-Wilk para determinar la distribución de las variables cuantitativas. Para las variables cuantitativas con distribución normal, se utilizó la media y la desviación estándar; y para las variables cuantitativas sin distribución normal, se utilizó la mediana y los rangos intercuartílicos.
El estudio fue aprobado por el Comité de Ética del Instituto Nacional de Salud del Niño CL-28-2017, contando además con la autorización administrativa institucional para el acceso a la información y el desarrollo del estudio.
HALLAZGOS
Se evaluaron 21 pacientes con diagnóstico clínico de agammaglobulinemia primaria. La edad media de los pacientes fue de 8,67 ± 4,25 años, 20/21 (95,2%) eran de género masculino, 9/21(42,9%) procedentes de provincias (Tabla 1).
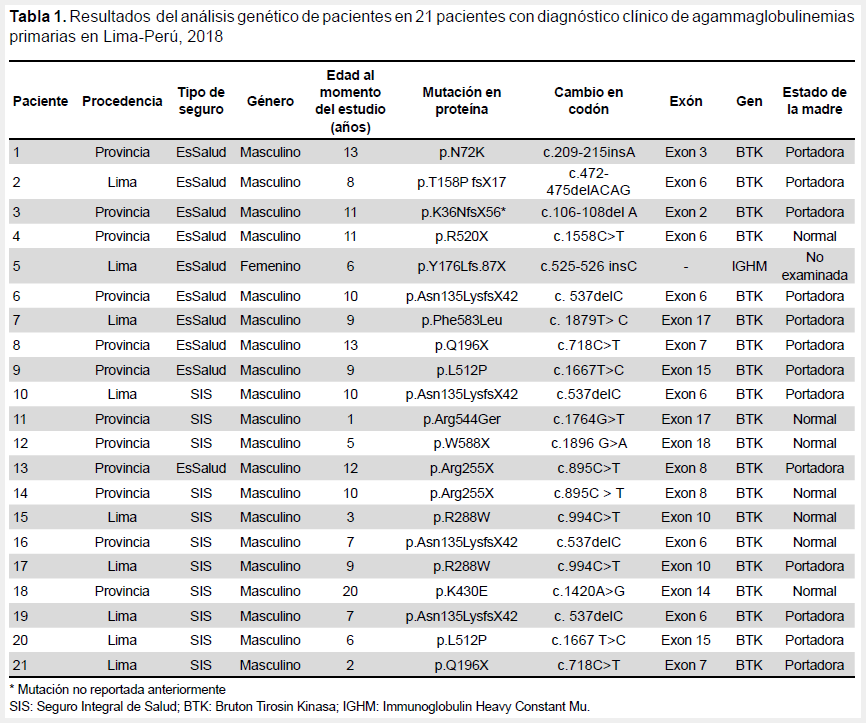
A todos los pacientes se les realizó el análisis genético, se encontró una mutación en la proteína p.K36NfsX56 y corresponde al gen BTK. El paciente actualmente se atiende en un hospital de EsSalud. En términos generales, encontramos 20 pacientes con mutaciones en el gen BTK y una mutación autosómica recesiva. De las mutaciones BTK encontramos 13 mutaciones hereditarias con una madre portadora y siete mutaciones de novo con una madre normal (Tabla 1). En una madre no se pudo realizar el análisis genético.
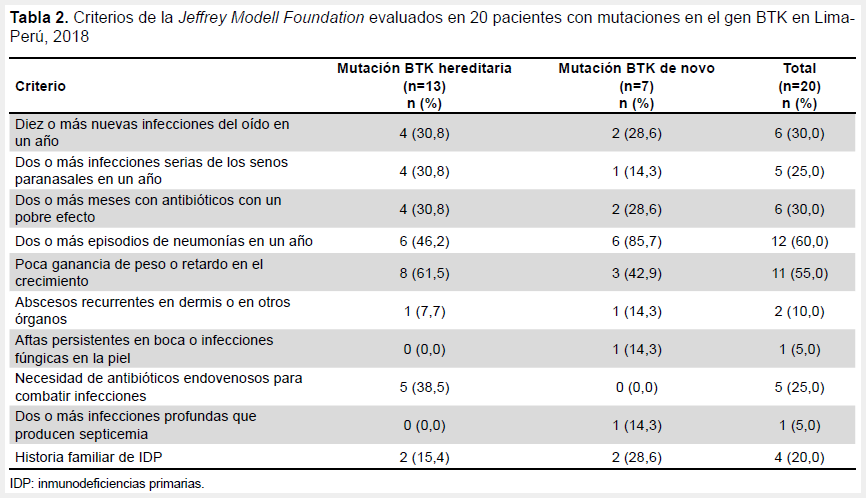
A todos los pacientes se les evaluó clínicamente en base a los criterios de la Jeffrey Modell Foundation. Los criterios que evidenciaron una mayor frecuencia fueron la ocurrencia de dos o más episodios de neumonías en un año con 12/20 (60,0%) de pacientes y la poca ganancia de peso o retardo en el crecimiento con 11/20 (55,0%) de pacientes (Tabla 2).
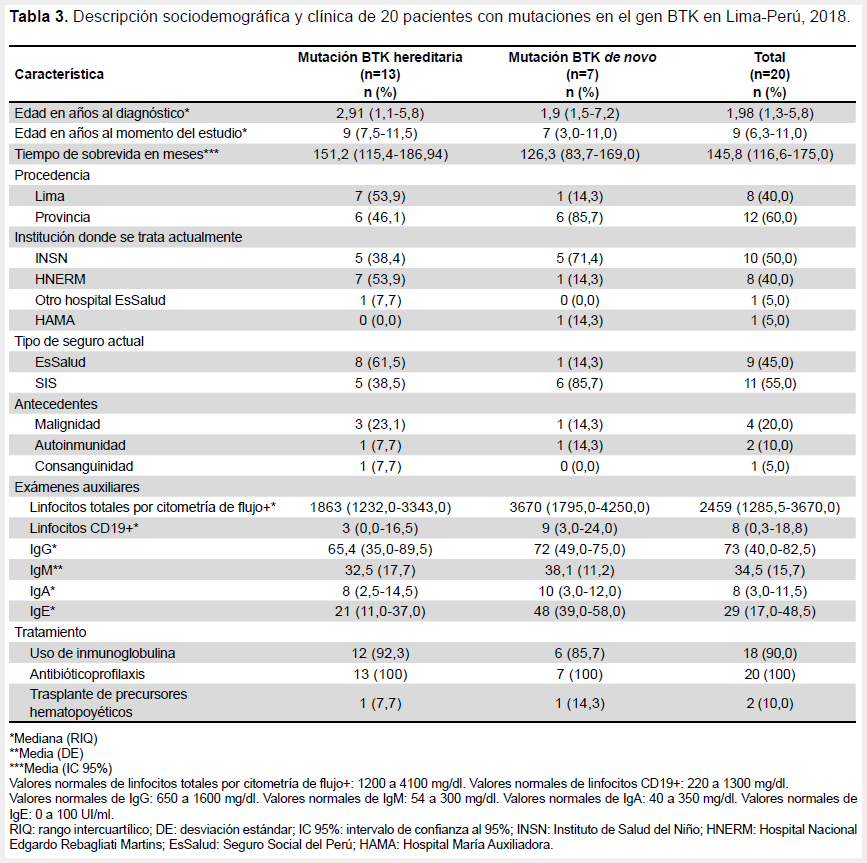
La Tabla 3 describe otros aspectos clínicos y terapéuticos en los pacientes con mutación BTK. Se encontró una mediana de linfocitos totales por citometría de flujo de 2459 con un rango intercuartílico entre 1285,5 y 3670. En cuanto al tratamiento, 18/20 (90,0%) de los pacientes recibieron inmunoglobulina y todos los pacientes recibieron antibiótico profilaxis. Sólo dos pacientes recibieron el tratamiento definitivo: trasplante de precursores hematopoyéticos.
DISCUSIÓN
Esta ha sido la primera aproximación en el Perú, al diagnóstico genotípico en pacientes con alta sospecha de AP. Todas las mutaciones encontradas han sido reportadas anteriormente, excepto la del gen BTK en la proteína p.K36NfsX56. Se reporta, a la vez, una mutación en el gen IGHM en la proteína p.Y176Lfs.87X. Cerca de un 60% de las mutaciones autosómico recesivas en AP se corresponden con alteraciones en la cadena pesada Mu, tal como lo reportó Silva et al. en el 2017 (11).
En relación con el tipo de mutación, se encontró que de las mutaciones BTK encontramos 13 mutaciones hereditarias y siete mutaciones de novo. En un estudio realizado en Turquía, las mutaciones del gen BTK se encontraron en el 69% de los casos y sólo tres fueron mutaciones de novo (12). En otro estudio realizado en China, se encontraron mutaciones en BTK en 16 de 21 pacientes y sólo 2 correspondían a mutaciones de novo (13). Las mutaciones de novo pueden influenciar sobre la severidad y la mortalidad de los pacientes, teniendo así, un impacto sobre el fenotipo clínico (14,15); sin embargo, en nuestro estudio no se encontró ninguna relación entre la severidad de la enfermedad y el tipo de mutación, y ello es acorde con los hallazgos de Esenbroga et al. quien manifiesta que no existe una relación entre el genotipo y el fenotipo de severidad de la enfermedad (8).
En la descripción clínica de los pacientes, se tiene que la edad mediana al diagnóstico fue de dos años. Ello se contrasta con un estudio realizado en el Perú en el 2017, Veramendi et al. encontró una edad media al diagnóstico de 7,4 años (16). Según datos previos, el 50,0% de los pacientes desarrolla manifestaciones clínicas al año de edad, y más del 95,0% desarrolla síntomas a los cinco años de edad; sin embargo, los síntomas desarrollados en edades tempranas suelen ser más severos e implican una mayor morbimortalidad (17). En consecuencia, la diferencia encontrada en cuanto a la edad de diagnóstico podría deberse a una mayor probabilidad de sospecha en el contexto de enfermedad severa o a la efectividad de los programas sobre capacitación en IDP que se vienen realizando desde los últimos años.
Las infecciones recurrentes estuvieron presentes en más del 60% de los casos y este antecedente constituye un aspecto clínico fundamental para establecer la sospecha diagnóstica de inmunodeficiencia primaria según la Jeffrey Modell Foundation (18). Se valoraron estos criterios en todos los pacientes incluidos en el estudio y se encontró que las neumonías considerando dos episodios en un año, y la pérdida de peso o retardo en el crecimiento fueron los eventos más frecuentes en los pacientes evaluados. Los microorganismos a los cuales un paciente con AP es más susceptible son los encapsulados S. pneumoniae y H. influenzae (3), en tal sentido, nuestros hallazgos expresan susceptibilidad probable a estas bacterias; sin embargo, no es posible determinar ello con precisión debido a que en dicho contexto no se toman cultivos de rutina.
El tratamiento definitivo de las AP es el trasplante de precursores hematopoyéticos (TPH); no obstante, las dificultades logísticas limitan la aplicación de esta terapia en nuestro medio, por lo que el tratamiento inicial de las AP se basa en la utilización de inmunoglobulina humana, así como en la instauración de antibióticoprofilaxis (17). Dos pacientes fueron sometidos al trasplante de precursores hematopoyéticos y ello se realizó fuera del país. Todos los pacientes recibieron inmunoglobulina. Cuando no es posible realizar un TPH, la utilización de inmunoglobulina es el único tratamiento capaz de alterar el curso natural de la enfermedad disminuyendo la morbilidad y la mortalidad (3). Más del 90% de los pacientes recibieron antibióticoprofilaxis. Además de la terapia de reemplazo de inmunoglobulina, los pacientes con ALX requieren tratamiento antibiótico agresivo para cualquier sospecha de infección y, en algunos casos, la terapia antibiótica prolongada puede estar indicada como tratamiento para infecciones pulmonares en curso o sinusitis crónica o como profilaxis (19).
Dado que el estudio tuvo una cantidad pequeña de pacientes, la posibilidad de realizar inferencias a partir de la información obtenida es limitada. Se debe considerar que aún existe un infradiagnóstico de pacientes con inmunodeficiencias primarias (9), por lo cual, el presente reporte es una primera aproximación a las AP y será necesario el registro continuo, además de contar con la logística necesaria para establecer el diagnóstico definitivo de AP en todos los casos que se puedan reportar. Sólo en dichas circunstancias, se podría reportar el perfil genotípico de los pacientes con AP además de sus implicancias clínicas y su impacto en el pronóstico.
En conclusión, el presente estudio muestra que las ALX son AP más frecuentes con mutaciones en los exones del gen BTK siendo estas mayoritariamente hereditarias, aunque existe un número elevado poco usual de mutaciones de novo. La presentación clínica habitual de las AP es con neumonías a repetición y la concurrencia de retardo en el crecimiento. Todos los pacientes del estudio recibieron inmunoglobulina humana, en tal sentido, es fundamental velar por la disponibilidad del fármaco en entornos de atención de pacientes con AP.
Contribuciones de autoría: EMB y DGG concibieron el proyecto de investigación. RIM y DGG elaboraron el proyecto de investigación hasta su aprobación. WCC, EMB y JAB participaron activamente en la revisión del proyecto hasta su aprobación. DGG realizó el análisis de datos. Todo el equipo participó en la elaboración del informe final.
Fuentes de financiamiento: autofinanciado.
Conflictos de interés: Los autores declaran no tener conflictos de interés.
REFERENCIAS BIBLIOGRÁFICAS
1. Ballow M. Primary immunodeficiency disorders: antibody deficiency. J Allergy Clin Immunol. 2002;109(4):581-91. doi: 10.1067/ mai.2002.122466.
2. Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova J, Chatila T, et al. The 2015 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2015;35(8):727-38. doi: 10.1007/s10875-015-0198-5
3. Howard V, Greene J, Pahwa S, Winkelstein JA, Boyle JM, Kocak M, et al. The health status and quality of life of adults with X-linked agammaglobulinemia. Clin Immunol Orlando Fla. 2006;118(2-3):201-8. doi: 10.1016/j.clim.2005.11.002
4. Gemayel K, Litman G, Sriaroon P. Autosomal recessive agammaglobulinemia associated with an IGLL1 gene missense mutation. Ann Allergy Asthma Immunol Off Publ Am Coll Allergy Asthma Immunol. 2016;117(4):439-41. doi: 10.1016/j.anai.2016.07.038
5. Tang P, Upton J, Barton M, Salvadori M, Clynick M, Price A, et al. Autosomal Recessive Agammaglobulinemia Due to a Homozygous Mutation in PIK3R1. J Clin Immunol. 2018;38(1):88-95. doi: 10.1007/s10875-017-0462-y
6. Winkelstein J, Marino M, Lederman H, Jones S, Sullivan K, Burks A, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85(4):193-202. doi: 10.1097/01.md.0000229482.27398.ad
7. Zhang X, Li H, Li Q, Gao J, Shi X. Detection of Bruton’s tyrosine kinase gene mutations and clinical analysis of 6 patients with X-linked agammaglobulinemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi Zhonghua Yixue Yichuanxue Zazhi Chin J Med Genet. 2014;31(1):29-33. doi: 10.3760/cma.j.issn.1003-9406.2014.01.007
8. Esenboga S, Cagdas D, Ozgur T, Gur P, Turkdemir L, Sanal O, et al. Clinical and genetic features of the patients with X-Linked agammaglobulinemia from Turkey: Single-centre experience. Scand J Immunol. 2018;87(3):e12647. doi: 10.1111/sji.12647
9. Mendoza D, García D, Córdova W. Diagnóstico situacional de las inmunodeficiencias primarias: aproximación preliminar. Rev Peru Med Exp Salud Publica. 2017;34(2):346. doi: 10.17843/rpmesp.2017.342.2687
10. García D, Córdova W, Aldave J. Registro de pacientes con inmunodeficiencias primarias en los tres principales centros de referencia del Perú. Rev Peru Med Exp Salud Publica. 2018;35(3):538. doi: 10.17843/rpmesp.2018.353.3317
11. Silva P, Justicia A, Regueiro A, Fariña S, Couselo J, Loidi L. Autosomal recessive agammaglobulinemia due to defect in μ heavy chain caused by a novel mutation in the IGHM gene. Genes Immun. 2017;18(3):197-9. doi: 10.1038/gene.2017.14
12. Wang Y, Kanegane H, Sanal O, Ersoy F, Tezcan I, Futatani T, et al. Bruton tyrosine kinase gene mutations in Turkish patients with presumed X-linked agammaglobulinemia. Hum Mutat. 2001;18(4):356. doi: 10.1002/humu.1200
13. Zhang Z, Zhao X, Jiang L, Liu E, Wang M, Yu J, et al. Clinical characteristics and molecular analysis of 21 Chinese children with congenital agammaglobulinemia. Scand J Immunol. 2010;72(5):454-9. doi: 10.1111/j.1365-3083.2010.02457.x
14. Abolhassani H, Vitali M, Lougaris V, Giliani S, Parvaneh N, Parvaneh L, et al. Cohort of Iranian Patients with Congenital Agammaglobulinemia: Mutation Analysis and Novel Gene Defects. Expert Rev Clin Immunol. 2016;12(4):479-86. doi: 10.1586/1744666X.2016.1139451
15. Acuna R, Veltman J, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 28 de 2016;17(1):241. doi: 10.1186/s13059-016-1110-1
16. Veramendi L, Zafra J, Pérez G, Córdova W. Diagnostic Delay of Primary Immunodeficiencies at a Tertiary Care Hospital in Peru- Brief Report. J Clin Immunol. 2017;37(4):383-7. doi: 10.1007/s10875-017-0398-2
17. Vivian H. Agammaglobulinemia In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA.
18. Modell V, Quinn J, Ginsberg G, Gladue R, Orange J, Modell F. Modeling strategy to identify patients with primary immunodeficiency utilizing risk management and outcome measurement. Immunol Res. 2017;65(3):713-20. doi: 10.1007/s12026-017-8907-1
19. Bonilla F, Khan D, Ballas Z, Chinen J, Frank M, Hsu J, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186-1205.e1-78. doi: 10.1016/j.jaci.2015.04.049.
Correspondencia: Edgar Emilio Matos Benavides.
Dirección: Av. Alejandro Bertello 855-Lima.
Teléfono: +51 995763350
Correo electrónico: ematben@hotmail.com
Recibido: 24/05/2019
Aprobado: 06/11/2019
En línea: 03/12/2019