10.17843/rpmesp.2020.372.4984
BRIEF REPORT
Molecular diversity in pathogenic variants of Vibrio parahaemolyticus in Peru
Junior Caro-Castro
![]() 1,2, Biologist, Bachelor in Microbiology and Parasitology
1,2, Biologist, Bachelor in Microbiology and Parasitology
Orson Mestanza ![]() 1, Biologist, Master in Bioinformatics
1, Biologist, Master in Bioinformatics
Willi Quino ![]() 1, Medical Technologist,
Master in Microbiology;
1, Medical Technologist,
Master in Microbiology;
Ronnie G. Gavilán
![]() 1,3, Biologist, Doctor in Biochemistry and Molecular Biology
1,3, Biologist, Doctor in Biochemistry and Molecular Biology
1 Instituto
Nacional de Salud, Lima, Perú.
2 Universidad Nacional Mayor de San Marcos, Lima, Perú.
3 Escuela Profesional de Medicina Humana, Universidad Privada San
Juan Bautista, Lima, Perú.
ABSTRACT
During the period from 1995 to 2017, in order to determine the diversity of Vibrio parahaemolyticus pathogenic variants in Peru, 102 Peruvian genomes (97 from a hospital setting and 5 from an out-of-hospital setting) were analyzed using the multilocus typification scheme and BLASTn in the search for virulence genes. Fifteen different sequence types were identified. It was found that the ST3 genotype, which is found in the pandemic clone, was the most abundant, with 52% (n=53); followed by ST120, with 23.5% (n=24); and the CC345 clonal complex, with 11.8% (n=12). A total of 89 analyzed strains presented genes encoding the pathogenicity island VpaI-7 (87.3%), while 96 presented the tdh gene (94.1%), and 6 the trh gene (5.9%). The ST3 genotype was the predominant one during the evaluated period, this genotype was the cause of a major outbreak in Peru’s past history. Other pathogenic genotypes found represent a latent public health risk associated with seafood consumption.
Keywords: Vibrio parahaemolyticus; Public Health, Epidemiological Monitoring; Molecular Typing; Whole Genome Sequencing (source: MeSH NLM).
INTRODUCTION
The presence of
pathogenic bacteria in the marine environment increases interest in food safety
because of their potential to cause outbreaks. Among them, the Vibrio parahaemolyticus stands out, a
halophilic gram-negative bacteria widely distributed in coastal
ecosystems, whose serotyping depends on somatic (O) and capsular (K) antigens
produced under various environmental conditions (1).
Interest
in V. parahaemolyticus began many years ago,
after it was found to be the causal agent of foodborne infections in an
outbreak in Japan. Historically, V. parahaemolyticus
has been responsible for 20-30% of cases of foodborne infection in Japan and
other Asian countries (2). Peru has recorded significant outbreaks
since 1997, which have been associated with climate changes that are part of
the El Niño phenomenon. These climate changes alter marine ecological
conditions, for example, increasing the rate of plankton abundance (3).
Most of these reports associate thermostable direct hemolysin
(TDH) with the virulence of V. parahaemolyticus
(4).
Currently,
the global prevalence and emergence of V. parahaemolyticus
infection is increasing, underlining the need for adequate surveillance of this
pathogen. Conventional microbiology is insufficient to determine pathogenic
variants and their geographical distribution. In contrast, molecular
epidemiology tools, such as the multilocus sequence
typing (MLST), are proposed as new alternatives to study infectious diseases
based on molecular strain differentiation. This scheme allows the rapid
genotypic characterization of microorganisms, due to the development of a
centralized database (PubMLST) that allows the
comparison of different genetic variants called sequence types (ST) and the
delineation of potential dispersion routes (5).
The
objective of this study was to determine the genetic variants of pathogenic
isolates of V. parahaemolyticus associated
with human cases and seafood circulating in Peru during 1995‑2017, using the
MLST technique and in silico detection of virulence genes.
|
KEY MESSAGES |
|
Motivation for the study: In view of the
possible global emergence of pathogens causing gastrointestinal infections,
strengthening molecular epidemiological surveillance of microorganisms such
as V. parahaemolyticus will contribute to
the timely detection and control of outbreaks. Main findings: Fifteen
different genotypes of V. parahaemolyticus
were detected, three of which have already caused important outbreaks in
Peru, while the other 12 have the potential to cause future epidemics due to
their virulent nature. Implications: To update
information on the circulating V. parahaemolyticus
genotypes in Peru until 2017, mainly prevalence and distribution through
time. |
THE STUDY
A total of 16 strains were submitted as V. parahaemolyticus from the collection of the National Reference Laboratory of Enteropathogens of the Instituto Nacional de Salud (INS) (Bioproject: PRJNA556706) and also 86 Peruvian genomes available in the NCBI database (http://www.ncbi.nlm.nih.gov) were included for MLST analysis (Table 1).
Table 1. Table of Peruvian Vibrio parahaemolyticus data used in this study.
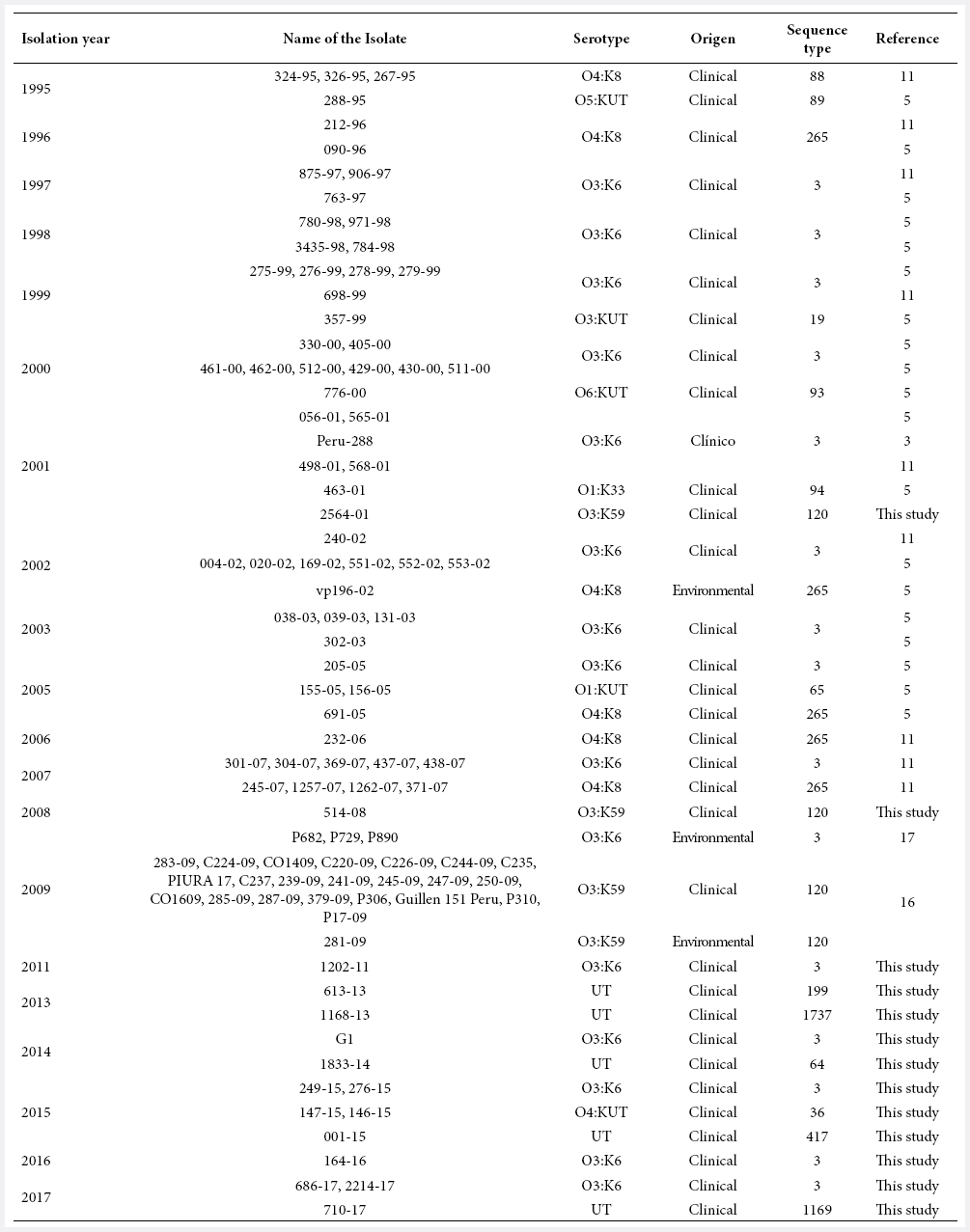
UT: Untypified.
The
strains were recovered in alkaline peptone water (Merck, Germany) at 37 °C
for 68 hours. Subsequently, they were seeded by striae
on bile-esculin citrate thiosulfate agar plates
(Merck, Germany), and incubated at 37 °C for 18 to 24 hours. The genus Vibrio
was confirmed using conventional biochemical tests,
and the species V. parahaemolyticus by PCR for
the presence of the toxR gene described by Kim
et al. (6).
DNA
extraction was performed using the DNeasy Blood
&Tissue kit (Qiagen, Germany). DNA concentration
and purity were evaluated by spectrophotometry (Denovix,
USA). Sequencing libraries were developed using the Nextera
XT kit (Illumina, USA), and genomic sequencing was performed using the MiSeq high throughput sequencer (Illumina, USA) (7).
The quality of the sequences obtained was evaluated using FastQC
v0.11.5. The sequences were assembled de novo using the A5-miseq
pipeline (8). Identification of the genus and detection of
contaminated contigs was carried out using the Kraken
program (9).
The
allelic profiles and the genomes’ STs obtained were assigned according to the
information in the MLST data base for V. parahaemolyticus
(http://pubmlst.org/vparahaemolyticus),
using the MLST v2.10 program, based on the seven-locus scheme described for V.
parahaemolyticus (5). Clonal
complex assignment was performed using BioNumerics
v7.5 (Applied Maths). Inclusion in a clonal complex
(CC) was restricted to STs that shared at least 6 of the 7 alleles, while
singletons were defined as STs that differed in two or more alleles from the
other STs. With the same program a minimum spanning tree (MST) was generated
showing the ST and the CC included in this work. In addition, a bar chart was
constructed from the isolates studied using Infostat,
to visualize genotypes by year of isolation and by number of samples.
The BLASTn tool was used to search for virulence factors of V.
parahaemolyticus: the pathogenicity island type 7
(VpI-7) that internally contains the most common variant of TDH obtained from
chromosome 2 of the RIMD genome 2210633 (access number: NC_004605.1) and TRH
obtained from isolate AQ4299 (access number: LC271586.1), identifying as
homologues those with <90% identity and <60% coverage of reference
alignment. The code used for the annotation is available at
http://github.com/OrsonMM/Blast-score-ratio-for-genomics.
The results obtained were ordered in table format indicating the presence or
absence of genes. All the sequences obtained during the study have been deposited
in GenBank (Bioproject
access number: PRJNA556706).
RESULTS
All 16 strains
were confirmed as V. parahaemolyticus due to
the presence of the ToxR gene. As for the
genomic information, an average genomic size of 5.18 bp
was obtained, composed by 80 contigs and a GC
percentage of 45.2%.
A total
of 102 genomes of Peruvian strains of clinical (97) and environmental (5)
origin, isolated during 1995-2017, were analyzed (Table 1). The 102 genomes
were classified into 15 different STs, which were grouped by year of isolation
and origin of the isolate (Figure 1). It is observed that no isolates were
obtained in 2004, 2010 and 2012.
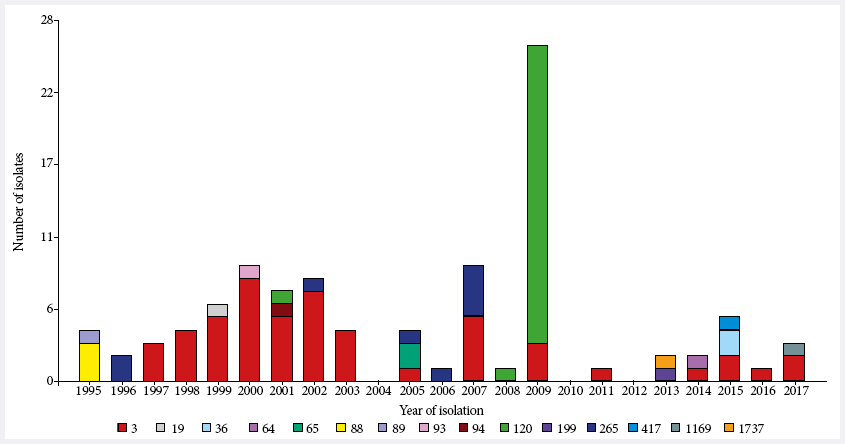
Figure 1. Distribution of Vibrio parahaemolyticus
by year of isolation, prepared with the InfoStat
program. The lower legend indicates the color according to the type of
sequence.
The populational structure of the Peruvian V. parahaemolyticus strains (n=102), analyzed by MLST, can
be visualized by means of a Minimum spanning tree graph (MST) (Figure 2). All
strains belonging to the O3:K6 pandemic complex were grouped in the ST3 (n=53),
representing 52% of the strains analyzed. In addition, clonal complex 345
(CC345) was identified, consisting of ST88 (n=3) and ST265 (n=9), both of
serotype O4:K8, which differ at a single locus, with an isolation frequency of
11.8%. The remaining strains were included in 12 unrelated STs, while ST120
23.5% (n=24) and ST36 1.9% (n=2) were notable for being related to local or
global outbreaks or epidemics (Annex 1).
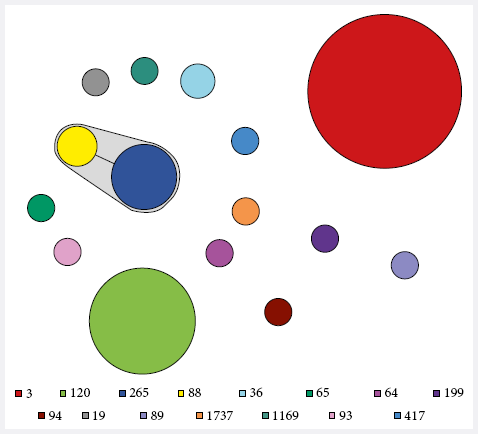
Figure 2. Minimum spanning tree (MST) of 102 V.
parahaemolyticus MLST allelic profiles
included in this study designed with BioNumerics v7.5
software. The caption indicates each type of sequence, differentiated by color.
Each circle represents a MLST genotype and the size is proportional to the
number of strains included in each one. The branches connecting the circles
indicate that they belong to the same clonal complex.
From
the 102 genomes analyzed, 89 isolates had the pathogenicity island, VpaI-7
(87.3%). Genes copies encoding TDH were found in 96
isolates (94.1%), the most frequent STs were ST3, ST36, ST88, ST120 and ST265.
Genes for TRH were found only in ST36, ST64, ST65 and ST417, for a total of 6
isolates (5.9%). The results grouped by ST can be seen in
Table 2, while the
results per gene isolates composing VpaI-7 can be seen in Annex 2.
Table 2. In silico detection of V. parahaemolyticus virulence factors by sequence type
|
Type of sequence |
n |
VpaI-7 |
tdhA |
tdhS |
trh |
|
ST3 |
53 |
53 |
53 |
53 |
0 |
|
ST19 |
1 |
0 |
0 |
0 |
0 |
|
ST36 |
2 |
0 |
2 |
2 |
2 |
|
ST64 |
1 |
0 |
1 |
1 |
1 |
|
ST65 |
2 |
0 |
0 |
0 |
2 |
|
ST88 |
3 |
3 |
2 |
2 |
0 |
|
ST89 |
1 |
0 |
1 |
1 |
0 |
|
ST93 |
1 |
0 |
1 |
1 |
0 |
|
ST94 |
1 |
0 |
0 |
0 |
0 |
|
ST120 |
24 |
24 |
23 |
23 |
0 |
|
ST199 |
1 |
0 |
1 |
1 |
0 |
|
ST265 |
9 |
9 |
9 |
9 |
0 |
|
ST417 |
1 |
0 |
1 |
1 |
1 |
|
ST1169 |
1 |
0 |
1 |
1 |
0 |
|
ST1737 |
1 |
0 |
1 |
1 |
0 |
|
Total |
102 |
89 |
96 |
96 |
6 |
n: number of isolates, VpaI-7: pathogenicity island 7, tdhA: thermostable direct haemolysin gene A, tdhS: thermostable direct haemolysin gene S, trh: TDH-related haemolysin gene
DISCUSSION
V. parahaemolyticus is a pathogen
transmitted by high-demand food, yet little information is available on
pathogenic variants and the temporal prevalence of its genotypes in Peru. This
under-report of V. parahaemolyticus infections
in the hospital setting is due to deficiencies in monitoring and research of
foodborne diseases (10).
When
analyzing the temporal prevalence of detected genotypes, the oldest isolates
are found to correspond to the years 1995-1996, especially serotype O4:K8, but
different genotypes: ST88 and ST265. Previous studies report this serotype
since 1980, which caused sporadic cases and small outbreaks associated with raw
seafood consumption; the highest prevalence was reported in 1983 (11).
Molecular surveillance studies revealed China as the origin of this serotype,
which is composed of several genotypes that have no clonal relationship with
the serotype O3:K6, suggesting CC345may be an
important clonal complex. In addition, comparative genomics analyses revealed
that these isolates presented the secretion system regions type 3 (T3SS) and
VpaI-7 (12), which is consistent with what was found in this study.
In the
distribution per year, the presence of ST3 stands out; and MST is the genotype
with the highest number of sequenced isolates, from strains isolated between
1997 and 2017. All of them had the complete VpaI-7. The first Peruvian outbreak
of ST3 occurred in 1997; however, it is known to have emerged in India in 1996,
expanding to the American continent (13). This genotype belongs to
the pandemic clonal complex CC3, which is distributed worldwide, and still is
the dominant clone today (5). This clonal complex is the most
studied genotype because it has the majority of VpaI
reported for V. parahaemolyticus, being VpaI-7
the most important one, associated to cytotoxicity and enterotoxicity
due to the presence of TDH and T3SS (14).
ST120
is the second group with the highest number of isolates in this study, obtained
mostly during 2009, due to a serotype O3:K59 outbreak in the northern regions
of the country (15). After the application of molecular surveillance
by MLST it was defined to be ST120, which originated from China, and represents
the third introduction of pathogenic populations of V. parahaemolyticus (16).
In the results, the presence of this genotype during 2001, many years before
its first report, is noteworthy, which would allow to reconsider when was the introduction of this genotype in Peru. Comparative
genomic studies would be necessary to find differences between these isolates
and those that caused the 2009 outbreak, which share the presence of VpaI-7.
Additionally,
ST36, a small group of epidemiological importance within the Peruvian isolates
analyzed, has its origin in the Pacific Northwest region of North America
causing outbreaks in the United States and Canada (1). It has
expanded since 2012 to rapidly reach other geographical areas such as
northwestern Spain (17). Based on our results, the first strains
belonging to this ST appeared in Peru in 2011, as part of the expansion of this
clone in the Pacific (18). Although no outbreak of this genotype has
been reported, it represents a latent epidemiological risk due to its
pathogenic potential because of the presence of TDH and TRH. In this aspect,
molecular surveillance by MLST can be applied for the timely tracking of these
isolates and to curb outbreaks.
No
information was found about other STs causing outbreaks or epidemics around the
world. However, their pathogenic potential is not ruled out, due to the
presence of genes encoding TDH or TRH, results that include genotypes such as
ST1169 and ST1737.
Virulence
factor analysis detected the presence of the genes encoding TDH in many
clinical isolates, particularly in the genotypes with the highest number of
isolates analyzed. It is known that the genes encoding TDH are mostly located
within VpaI-7, so the presence of this region will have an impact on the
increase in virulence of V. parahaemolyticus (19).
However, the genetic deletion of the copies of the tdh
gene or of the complete VpaI-7 does not determine the absence of virulence (14),
which explains the detection of clinical strains with absence of TDH. On the
other hand, genomes with TDH but without VpaI-7 were detected, which had
already been described as TDH variants not associated with VpaI-7 (20).
Finally, the gene encoding TRH, which causes a similar effect to TDH, was found
in a very small group of isolates.
In conclusion, ST3 is both temporally and quantitatively predominant in Peru, that is why still today, it is a genotype that generates risk in public health associated with the consumption of raw or semi-raw seafood. Notably, its pathogenic potential is due to the presence of VpaI-7, carrier of hemolysins. This, added to the underestimated epidemiological data, as well as the circulation of other pathogenic variants of these bacteria in the country, indicates that bigger efforts in molecular surveillance are needed, because this method is proving to be a powerful tool to detect and control outbreaks and infections.
Acknowledgements:
REFERENCES
1. Banerjee SK, Kearney AK, Nadon CA, Peterson C-L, Tyler K, Bakouche L, et al. Phenotypic and Genotypic Characterization of Canadian Clinical Isolates of Vibrio parahaemolyticus Collected from 2000 to 2009. J Clin Microbiol. 2014;52(4):1081-8. doi: 10.1128/JCM.03047-13.
2. Alam MJ, Tomochika KI, Miyoshi SI, Shinoda S. Environmental investigation of potentially pathogenic Vibrio parahaemolyticus in the Seto-Inland Sea, Japan. FEMS Microbiol Lett. 2002;208(1):83-7. doi: 10.1111/j.1574-6968.2002.tb11064.x.
3. Aliaga R, Miranda J, Zevallos J. Aislamiento e identificación de Vibrio parahaemolyticus O3: K6 en pescados y moluscos bivalvos procedentes de un mercado pesquero de Lima, Perú. Rev Medica Hered. 2010;21(3):139-45. doi: 10.20453/rmh.v21i3.1123.
4. Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global Dissemination of Vibrio parahaemolyticus Serotype O3:K6 and Its Serovariants. Clin Microbiol Rev. 2007;20(1):39-48. doi: 10.1128/CMR.00025-06.
5. Gonzalez-Escalona N, Jolley KA, Reed E, Martinez-Urtaza J. Defining a Core Genome Multilocus Sequence Typing Scheme for the Global Epidemiology of Vibrio parahaemolyticus. J Clin Microbiol. 2017;55(6):1682-97. doi: 10.1128/JCM.00227-17.
6. Kim YB, Okuda J, Matsumoto C, Takahashi N, Hashimoto S, Nishibuchi M. Identification of Vibrio parahaemolyticus strains at the species level by PCR targeted to the toxR gene. J Clin Microbiol. 1999;37(4):1173-7.
7. Quino W, Hurtado CV, Escalante-Maldonado O, Flores-León D, Mestanza O, Vences-Rosales F, et al. Multidrogorresistencia de Salmonella infantis en Perú: un estudio mediante secuenciamiento de nueva generación. Rev Peru Med Exp Salud Pública. 2019;36(1):37-45. doi: 10.17843/rpmesp.2019.361.3934.
8. Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinforma Oxf Engl. 2015;31(4):587-9. doi: 10.1093/bioinformatics/btu661.
9. Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46. doi: 10.1186/gb-2014-15-3-r46.
10. Martinez-Urtaza J, Lozano-Leon A, DePaola A, Ishibashi M, Shimada K, Nishibuchi M, et al. Characterization of Pathogenic Vibrio parahaemolyticus Isolates from Clinical Sources in Spain and Comparison with Asian and North American Pandemic Isolates. J Clin Microbiol. 2004;42(10): 4672–78. doi: 10.1128/JCM.42.10.4672-4678.2004.
11. Gavilan RG, Zamudio ML, Martinez-Urtaza J. Molecular epidemiology and genetic variation of pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl Trop Dis. 2013;7(5):e2210. doi: 10.1371/journal.pntd.0002210.
12. Li B, Yang X, Tan H, Ke B, He D, Ke C, et al. Vibrio parahaemolyticus O4:K8 forms a potential predominant clone in southern China as detected by whole-genome sequence analysis. Int J Food Microbiol. 2017;244:90-5. doi: 10.1016/j.ijfoodmicro.2017.01.001.
13. Guerrero A, Lizárraga-Partida ML, Gil BG, Licea-Navarro AF, Revilla-Castellanos VJ, Wong-Chang I, et al. Genetic Analysis of Vibrio parahaemolyticus O3:K6 Strains That Have Been Isolated in Mexico Since 1998. PLoS ONE. 2017;12(1): e0169722. doi: 10.1371/journal.pone.0169722.
14. Ceccarelli D, Hasan NA, Huq A, Colwell RR. Distribution and dynamics of epidemic and pandemic Vibrio parahaemolyticus virulence factors. Front Cell Infect Microbiol . 2013;3:97. doi: 10.3389/fcimb.2013.00097.
15. Zamudio ML, Meza A, Bailón H, Martinez-Urtaza J, Campos J. Experiences in the epidemiological surveillance of foodborne pathogens by pulsed field gel electrophoresis (PFGE) in Peru. Rev Peru Med Exp Salud Publica. 2011;28(1):128-35.
16. Gonzalez-Escalona N, Gavilan RG, Toro M, Zamudio ML, Martinez-Urtaza J. Outbreak of Vibrio parahaemolyticus Sequence Type 120, Peru, 2009. Emerg Infect Dis. 2016;22(7):1235-7. doi: 10.3201/eid2207.151896.
17. Martinez-Urtaza J, Aerle RV, Marin MA, Haendiges J, Myers RA, Trinanes J, et al. Genomic variation and evolution of vibrio parahaemolyticus ST36 over the course of a transcontinental epidemic expansion. mBio. 2017;8(6):e01425-17. doi: 10.1128/mBio.01425-17.
18. Abanto M, Gavilan RG, Baker-Austin C, Gonzalez-Escalona N, Martinez-Urtaza J. Global Expansion of Pacific Northwest Vibrio parahaemolyticus Sequence Type 36. Emerging Infectious Diseases. 2020;26(2):323-6. doi: 10.3201/eid2602.190362.
19. Raghunath P. Roles of thermostable direct hemolysin (TDH) and TDH-related hemolysin (TRH) in Vibrio parahaemolyticus. Front Microbiol. 2015;5:805. doi: 10.3389/fmicb.2014.00805.
20. Okada N, Iida T, Park KS, Goto N, Yasunaga T, Hiyoshi H, et al. Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infection and immunity. 2009;77(2):904–13. doi: 10.1128/IAI.01184-08.
Correspondence to: Junior
Caro Castro; Laboratorio de Referencia
Nacional de Enteropatógenos, Instituto
Nacional de Salud, Cápac Yupanqui 1400, Jesús María, Lima, Perú;
juniorcaro12@hotmail.com.
Authors’
contribution: JC, RG and WQ participated in the conception,
hypothesis delineation and study design. JC and OM participated in the
analysis, data interpretation and writing of the article. RG and OM
participated in the critical review of the article. All approved the final
version.
Conflicts of
interest: None.
Funding sources: The
research was funded by Cienciactiva/FONDECYT
(Agreement 145-2017-FONDECYT) and by the Instituto
Nacional de Salud, Lima, Peru (OGITT: OI-0037-17).
Citation: Caro-Castro J, Mestanza
O, Quino W, Gavilán RG. Molecular diversity in pathogenic variants of Vibrio parahaemolyticus in Peru. Rev Peru Med Exp Salud Pública.
2020;37(2):270-5. doi: https://doi.org/10.17843/rpmesp.2020.372.4984.
Received: 20/11/2019
Approved: 29/04/2020
Online: 01/06/2020